Soil metagenomes from greenhouse study with biofertilizer
Overview
We are analyzing the metagenomes from 2 soil samples that were used a greenhouse study prior to biofertilizer treatment. These will give us a sense of resident microbial (fungal and bacterial) communities residing in them.
- F1B-KM40 - bulk field soil
- F2R-KM41 - rhizosphere soil
The goal is to generate phylogenetic trees of bacteria and fungi in these samples.
Methods
- Confirm data integrity with checksums
- Quality control: Fast QC and MultiQC
- Preprocessing: trimming via Trimmomatic, merging with PEAR
- Assembly via Megahit
- Tree construction using Megan
Trimming
Trimming removes adapters and low-quality bases.
This produces unpaired and paired file outputs. Paired reads are those which both forward and reverse survived trimming. These are used for downstream analysis like merging and assembly. Unpaired reads indicate where only one of the pair survived (the other was discarded due to low quality or short length).
Merging
These are R1/R2 (forward and reverse reads) and will have to be merged. This is the last component of pre-processing as we work towards metagenome assembly.
/home/shared/fastp-v0.24.0/fastp \
-i F1B-KM40_trimmed_R1_paired.fastq.gz \
-I F1B-KM40_trimmed_R2_paired.fastq.gz \
--merge \
--merged_out F1B-KM40_merged.fastq.gz \
/home/shared/fastp-v0.24.0/fastp \
-i F2R-KM41_trimmed_R1_paired.fastq.gz \
-I F2R-KM41_trimmed_R2_paired.fastq.gz \
--merge \
--merged_out F2R-KM41_merged.fastq.gz Assembly
To assemble the metagenome files, MEGAHIT was used.
./megahit
-r ../F1B-KM40_merged.fastq.gz #specifying input file
-o megahit_F1B_KM40_out #output directory
--min-contig-len 500 #over 500 bps
-t 8 #8 threadsLike other steps, this is done with both files.
Results
Phylogenetic trees are constructed using Megan. This is the result for F2R, the rhizosphere soil. The code and results for F1B will be included in the next update.
Rhizosphere soil top hits
| Organism | Classification |
|---|---|
| Acidobacteria bacterium | Bacteria |
| Alphaproteobacteria bacterium | Bacteria |
| Betaproteobacteria bacterium | Bacteria |
| Verrucomicrobia bacterium | Bacteria |
| Actinobacteria bacterium | Bacteria |
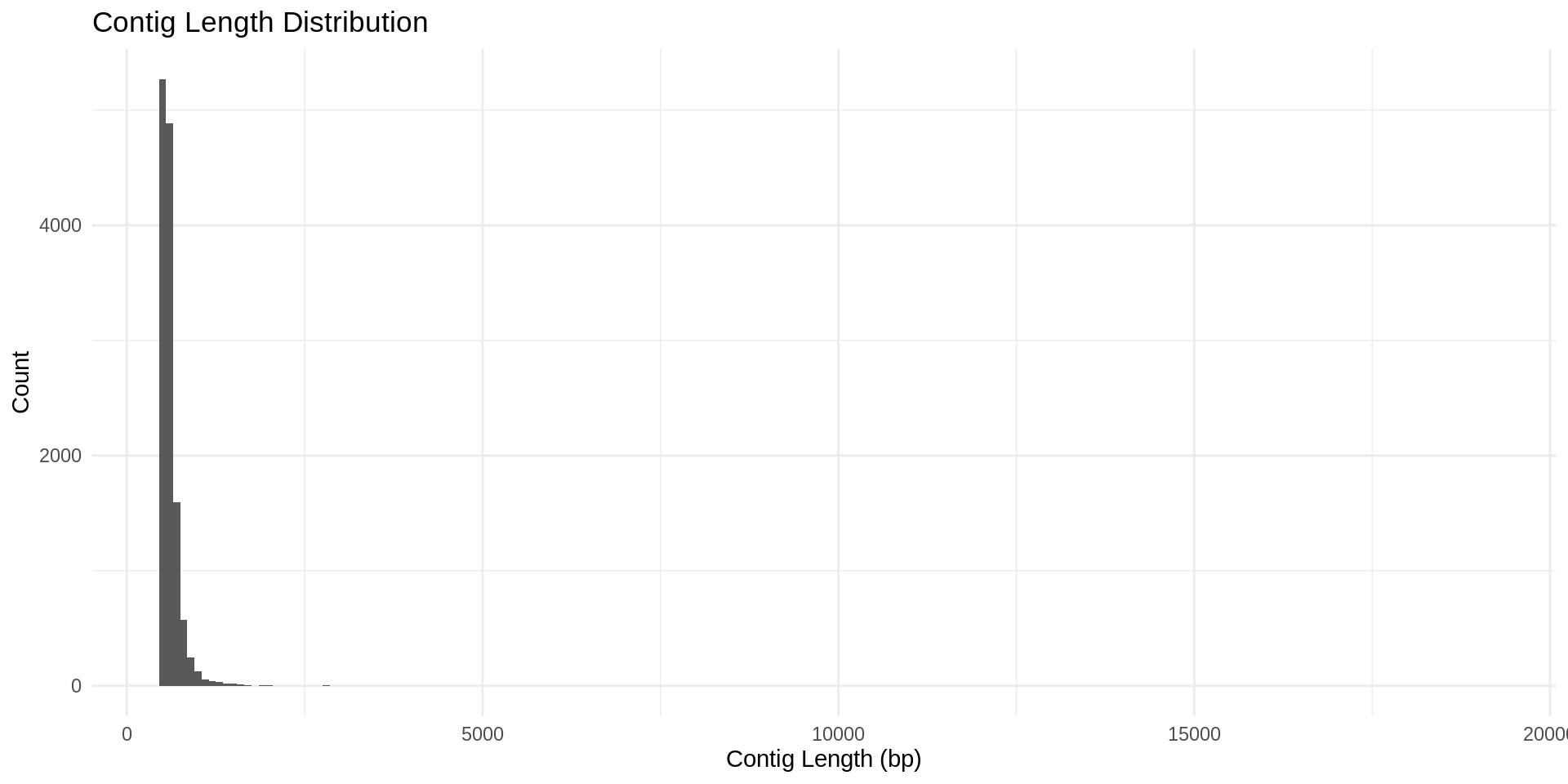
Plot: Contig length
To better visualize our results, we will plot a histogram of contig lengths for this F2R rhizosphere soil sample.
Next 4 weeks
Taxonomy for F1B (bulk soil)
Annotation via MG-RAST
Visualizations
Plan for remaining 46 metagenomes after course completion