Week 06 Project Updates
Liz Rockfish Updates
Welcome to a tale of many trials and few triumphs!
Here is a picture of a copper rockfish to get the energy up:

I mean, just look at it!!
Objective of this project
I will be taking RAD-seq data from three species of closely-related rockfish species (Sebastes spp.) through a bioinformatics processing pipeline and producing a PCA plot that displays population structure clusters.
Steps in the pipeline
- Run QC on raw sequence data using FastQC and interactively visualizing with MultiQC
- Demultiplex to the individual sample level using the STACKS
process_radtagsprogram and remove PCR duplicates withclone_filter - Align to the honeycomb rockfish genome (the closest relative that has a reference-quality genome assembly) using
bowtie2 - Use
gstacksandpopulationsmodules in STACKS to perform SNP calling and basic statistics - Create PCA plots using
adegenet, an R package, to examine inter- and intraspecific population structure patterns & clustering
MultiQC results
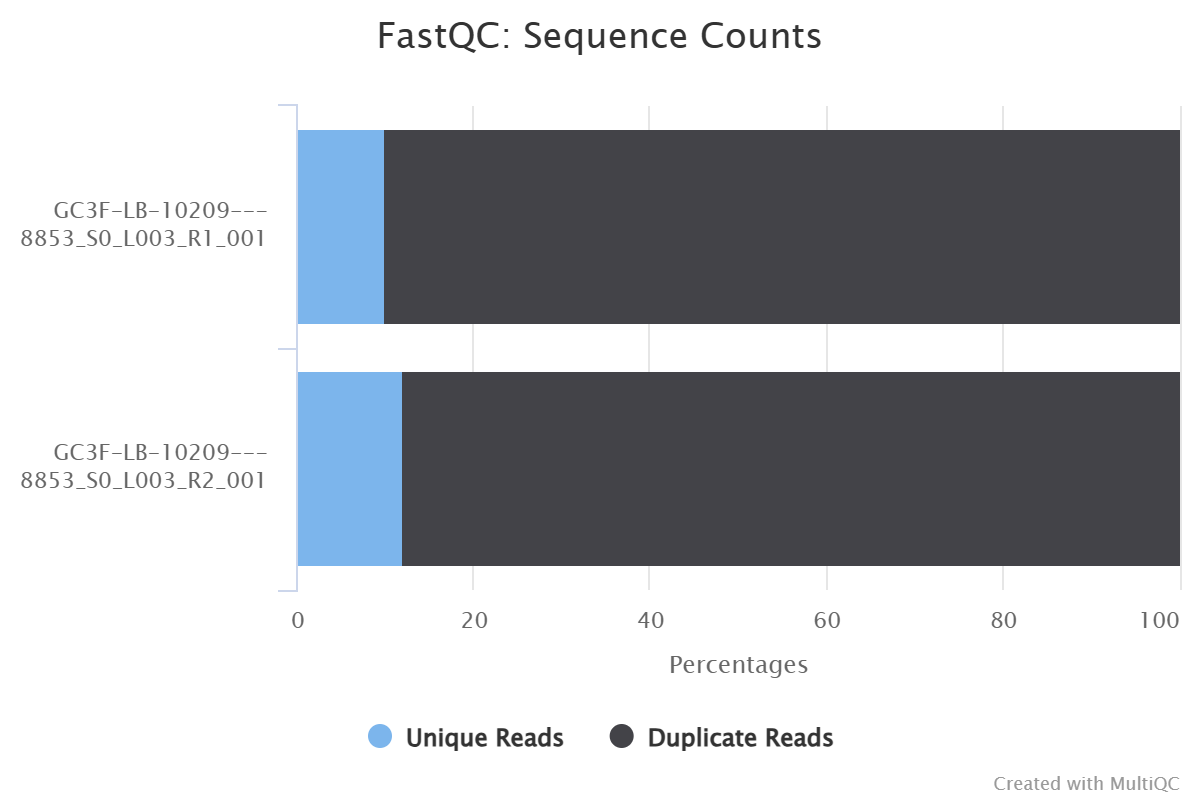
FastQC sequence counts
MultiQC results continued
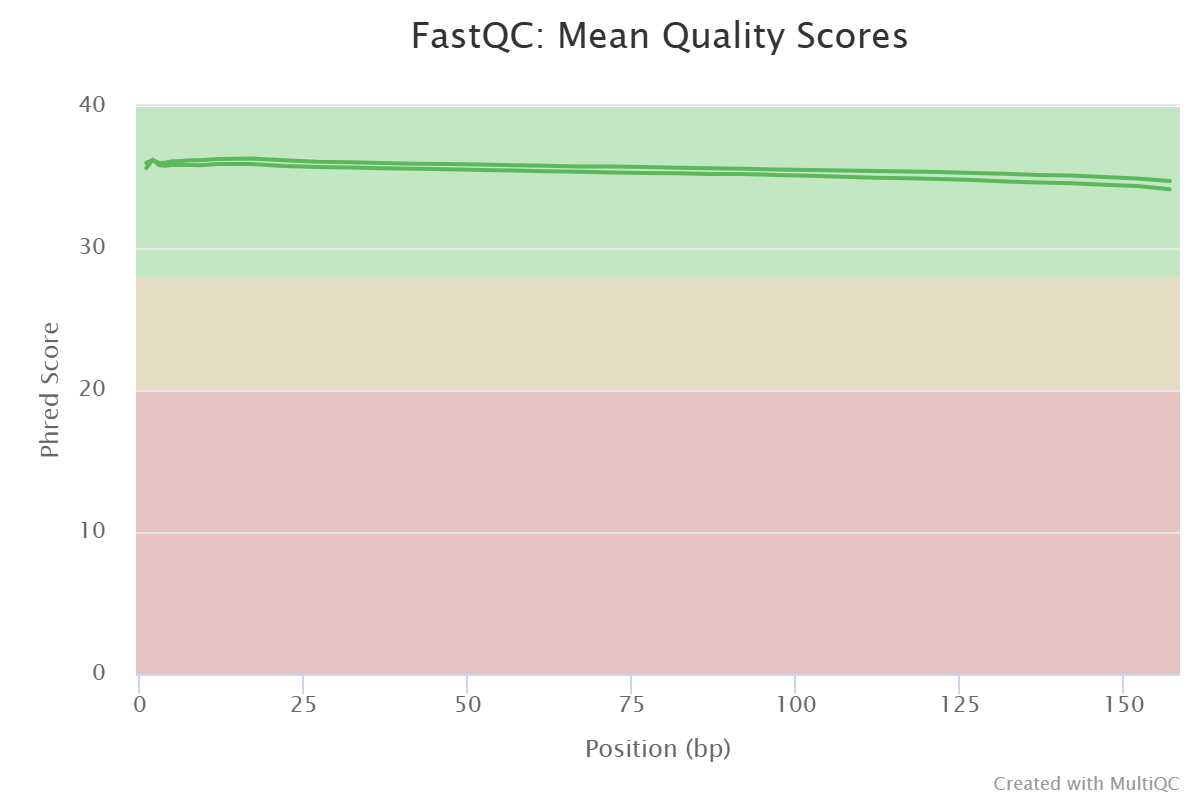
FastQC sequence quality
Demultiplexing: disaster?
Demultiplexing is normally a pretty standard step in the pipeline, but my data has decided to send me on a journey instead :)
So I will be sharing where I’m at and how I got there!
A look at my data
Here is what a peek at my raw sequence file - R1, or the forward read - looks like using the head command:
[lizboggs@klone-login03 radseq_data]$ zcat GC3F-LB-10209---8853_S0_L003_R1_001.fastq.gz | head
@A01335:621:H5WGLDSXF:3:1101:1036:1000 1:N:0:GCCAAT+AGATCT
GGACAGCAGATGCAGGCATGCGCCATGGAGATTAGCTACACCGCAGTGGCGTGGAACACATAACAGGCTCATTAGGTAGCATATTCTCCAGGAGGCATGCGCCTATGGAGAATAGCCACACCGCAGTGGCGTGGAACACATAACAGGCTCACTAGGTAT
+
FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF,FFFFFFFF:FFF,FFFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:F:FFF,FFFFFF:FFFFFFFFFFFFFF:,FFFFFFFFFFFF:FF::,
@A01335:621:H5WGLDSXF:3:1101:1072:1000 1:N:0:CGATGT+AGATCT
CGGAGGAGCAGGTGTTGATGGGGCCGTTCTTCATCTGCTCCAGAGTGTCCAGCACCACGTCGGGAGCCGGCCGGCAGCCCTGCCGCTCCACCTGCCTCAACTCGCCCACAGCCACCGTCACCGTCACCTCGAAGTCCTGCATGTCTATCCCAGATCGGA
+
F:FFFFF:F:FFFF,::F:FF:,F,FFF::F:FF:F,F,::FFFF,,F,FF,FFFFF:,FFFFFF,FF:FF:,:FF,FFF,::F,F,:F:,FF:FF,:FFF,,,,FF,,F,:::::,,FFFF,,:,:F::,F,:,F,,,F:,:F:,F,,FFFFFF,,,,
@A01335:621:H5WGLDSXF:3:1101:1090:1000 1:N:0:ATCACG+AGATCT
CCACCACACAGCCGCCCTCCAGCTCACCACGGCCCTCAGCCTCTTCACGAGCTGCAACCACCACACAGCTGCCCTCCACCTCACCACGGCCCTCAGCCTCTTTGCGAGCTGCCCCCTCCACACAGCTGCTCTCCACCTCACCACGGCCCTCAGCCTCTT
The first line is the sequence header (unique to each read), second line is the read itself, third is a + separator line, and fourth is a Phred quality score for each base
A look at my code
Here is the script I am using to run STACKS process_radtags on my data:
#!/bin/bash
#SBATCH --job-name=process_radtags_rfnewbarcode_lib5_712fix
#SBATCH --account=merlab
#SBATCH --partition=compute-hugemem
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=32
#SBATCH --time=2-16:00:00
#SBATCH --mem=500G
#SBATCH --mail-type=ALL
#SBATCH --mail-user=lizboggs@uw.edu
##### ENVIRONMENT SETUP ##########
module purge
source /gscratch/merlab/software/miniconda3/etc/profile.d/conda.sh
conda activate stacks_env
# Set paths
RAWDIR=/mmfs1/gscratch/merlab/lizboggs/radseq_data
OUTDIR=/mmfs1/gscratch/scrubbed/lizboggs/newbarcodedemux_lib5indx712
BARCODE_DIR=/mmfs1/gscratch/merlab/lizboggs/barcodes
process_radtags \
-1 ${RAWDIR}/GC3F-LB-10209---8853_S0_L003_R1_001.fastq.gz \
-2 ${RAWDIR}/GC3F-LB-10209---8853_S0_L003_R2_001.fastq.gz \
-b ${BARCODE_DIR}/allbarcodes_new_withlib5indx712fix.txt \
-o ${OUTDIR} \
--renz_1 sbfI \
--inline_index \
-i gzfastq \
-y fastq \
-E phred33 \
--bestrad \
-q \
--filter_illumina \
-c \
-t 130 \
--adapter_1 AGATCGGAAGAGCACACGTCTGAACTCCAGTCA \
--adapter_2 AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT \
-r \
--barcode_dist_1 1 \
--barcode_dist_2 1
# Close environment
conda deactivateThe output from this code (after a 63.5-hour job run!)
6228064294 total reads; -0 failed Illumina reads; -3580073710 ambiguous barcodes; -28122894 ambiguous RAD-Tags; +83758324 recovered; -5554816 low quality reads; 2563948807 retained reads.
50364067 reads with adapter sequence.
Closing files, flushing buffers...done.
6228064294 total sequences
1494217429 reads were transposed [based on the BestRAD flag] (24.0%)
0 failed Illumina filtered reads (0.0%)
50364067 reads contained adapter sequence (0.8%)
3580073710 barcode not found drops (57.5%)
5554816 low quality read drops (0.1%)
28122894 RAD cutsite not found drops (0.5%)
2563948807 retained reads (41.2%)
Only 41.2% of reads retained and 57.5% “barcode not found” drops… something is up.
THE LIKELY CULPRIT: the barcodes
Some important background: reads are demultiplexed based on their indexes. In order for process_radtags to know which reads are what, it needs to be told where to find the indexes.
This is done with program flags - in my case, I used the flag --inline_index, as my 10 base-pair individual barcodes are inline, ie. they are found in the actual read. My library index - ie. which library the individual belonged to - is found in the header.
I’ll show on the next slide why this organization is causing an issue in my processing.
Index mix-up
When using the command grep -A 20 "barcodes_not_recorded" process_radtags.radseq_data.log, which pulls all “barcodes not recorded” (ie. dropped), it reports this:
BEGIN barcodes_not_recorded
# A list and count of barcodes found that were not specified in the barcodes input file.
Barcode Total
GGGAGCTGAA-AGATCT 104988366
GGAAACATCG-AGATCT 88023132
GGCCGTGAGA-AGATCT 55435788
GGGACAGTGC-AGATCT 52357626
GGCGAACTTA-AGATCT 51776254
GGACCACTGT-AGATCT 50442810
GGGATAGACA-AGATCT 50208624
GGCATCAAGT-AGATCT 46791976
GGAACGCTTA-AGATCT 44114520
GGATCCTGTA-AGATCT 44070132
GGTCCGTCTA-AGATCT 42453748
GGAGAGTCAA-AGATCT 40700258
GGCCTCCTGA-AGATCT 40518226
GGCGCATACA-AGATCT 40145892
GGTGGTGGTA-AGATCT 38694062
GGCAAGACTA-AGATCT 38661102
GGTGAAGAGA-AGATCT 37881860
GGAATCCGTC-AGATCT 37656988
--
END barcodes_not_recordedWhy that’s a problem
It seems to be matching a handful of the 10bp individual barcodes with the 6bp i5 Illumina barcode that is on every read, and thus… it can’t assign those reads to a single individual, because 4-5 individuals will have both of those barcodes attached to them.
Reviewing this section of my R1 file:
@A01335:621:H5WGLDSXF:3:1101:1036:1000 1:N:0:GCCAAT+AGATCT
GGACAGCAGATGCAGGCATGCGCCATGGAGATTAGCTACACCGCAGTGGCGTGGAACACATAACAGGCTCATTAGGTAGCATATTCTCCAGGAGGCATGCGCCTATGGAGAATAGCCACACCGCAGTGGCGTGGAACACATAACAGGCTCACTAGGTATThe library index is the first in the index+index pair; here it is GCCAAT, which is the index for my library 5. The second in that pair is that Illumina i5 index that is on every read. The 10bp individual barcode is the first 10bp of the actual read. So while process_radtags is correctly reading at least 40% of reads correctly - pulling the first index barcode and pairing it with its respective individual barcode - it has also paired the wrong ones together and doesn’t know what to do with them, so it drops them as “not found.”
What’s next
1. Cry (check! Just kidding… maybe)
Field as much help as possible, from lab mates to the internet, ie. Bioinformatics Stack Exchange (check!)
Contact the sequencing facility and make it their problem by sending my data back for THEM to demux (very close to doing this…)
Once data is finally demux’d in whatever way… continue with pipeline!!!
In the meantime…
I still need to prove my proficiency in making a Quarto presentation, so let’s do some other fun things with my metadata instead
Check it out! A table of samples!
| Species | # of Samples |
|---|---|
| Brown | 97 |
| Copper | 181 |
| Quillback | 217 |
This is in total, accounting for combining my rockfish with those from a previous study (not done by me). Lots of fish!
Plot time
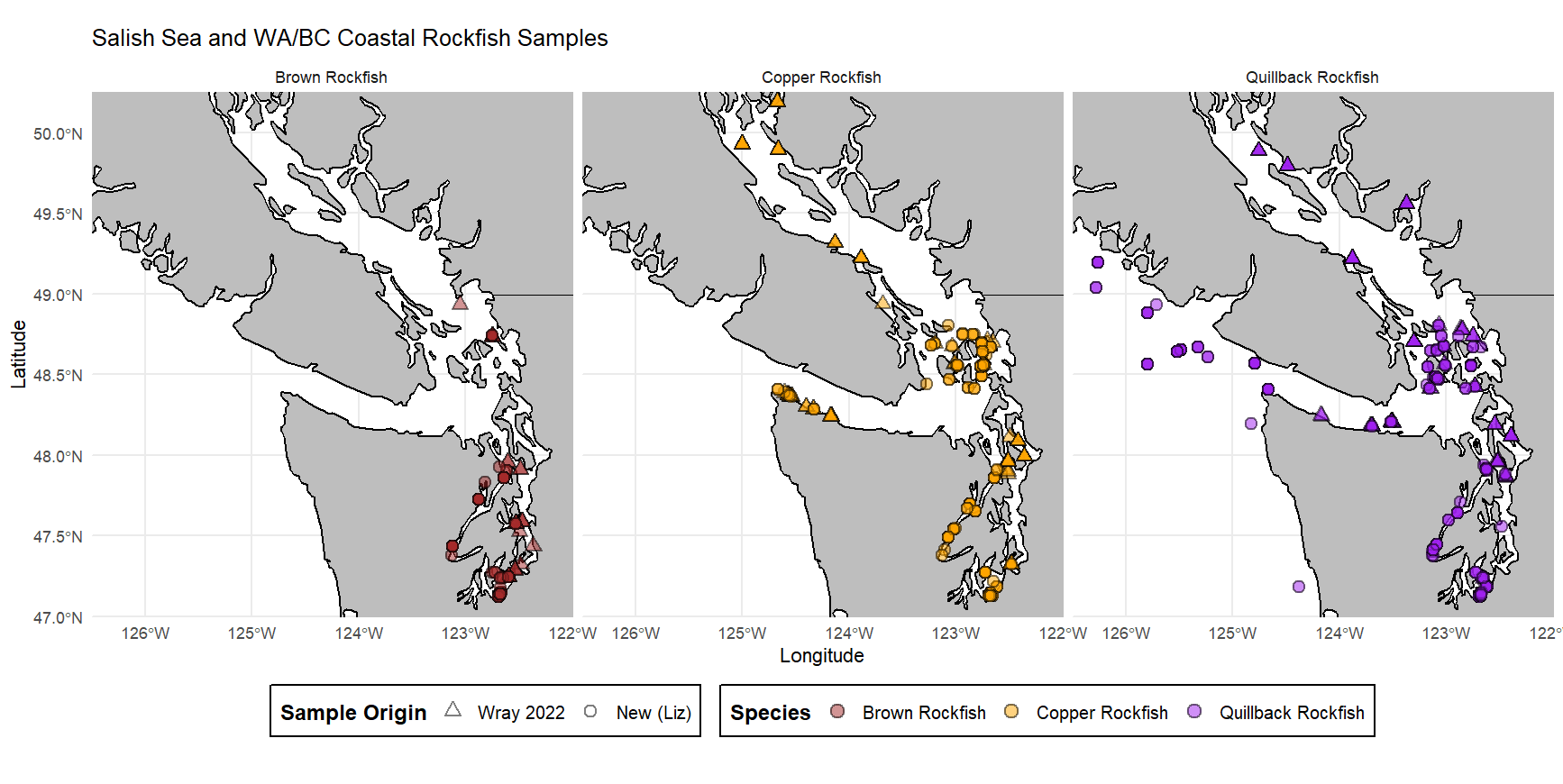
Plot of my rockfish samples by species!
That’s all for now!
I hope this demonstrated my ability to work with Quarto and showed off my very neat very fun coding adventure… hopefully have a good result soon!