Creating a workflow for genome-environment-association analyses with RADseq data
Lara Breitkreutz
Project Goal
create a workflow for conducting genotype-environment association (GEA) analyses using RAD-seq data
in the future, I will use this workflow for identifying candidate loci associated with components of thermal variation
Methods: alignment
- Aligned sequences to a reference Zostera marina genome using the BWA MEM function in the Burrow-Wheeler Aligner software (Li and Durbin, 2009).
Methods: alignment
## Aligning
# a for loop that aligns each .fq file according to name of sample and outputs a .sam file
for i in ${ind_ID};
do
bwa mem -t 20 -T 30 $REF $INPUT/${i}.fq > $OUTPUT1/${i}.sam 2>> $REPORT
done
conda deactivate #deactivate conda env
## Sorting
conda activate samtools_env
# a for loop that sorts each .sam file (using samtools) and creates a .bam file
for i in ${ind_ID};
do
samtools view -b -q 30 $OUTPUT1/${i}.sam > $OUTPUT1/${i}.bam
samtools sort $OUTPUT1/${i}.bam > $OUTPUT2/${i}.bam
samtools index $OUTPUT2/${i}.bamMethods: genotyping
- Genotyping polymorphic loci using the ref_map function in STACKS.
- Important input file for this script is the text file that stores the samples and their corresponding collection site and region
Methods: genotyping
| sample | collection site | region |
|---|---|---|
| B001 | Belfair | HoodCanal |
| B002 | Belfair | HoodCanal |
| … | … | … |
| W049 | Willapa Bay | Coast |
| W050 | Willapa Bay | Coast |
Methods: genotyping
Methods: environmental data simulation
- Awaiting arrival of environmental data I have requested (and request has been granted)
- In the mean time, simulating some…
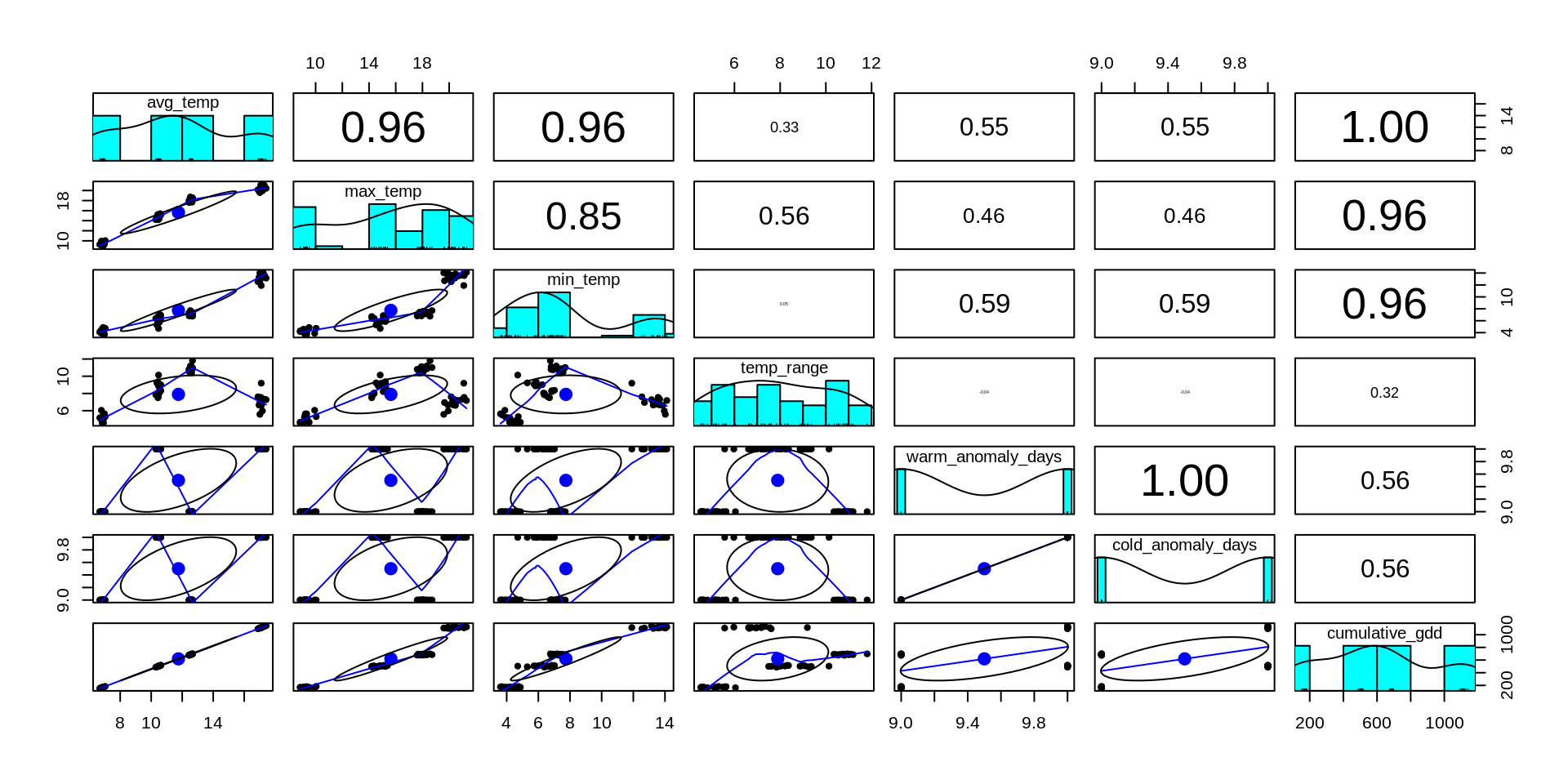
Methods: environmental data simulation
# function for simulating temperatures
simulate_temps <- function() {
base_temps <- c(
6.5, 7.1, 8.3, 10.2, 12.8, 15.5, # Monthly averages for nearshore areas
17.5, 18.3, 16.1, 12.5, 9.1, 7.2 # Based on Salish Sea observations
)
tibble(date = seq.Date(as.Date("2025-01-01"), by = "day", length.out = 365)) %>%
mutate(
month = month(date),
temp = base_temps[month] + rnorm(365, 0, 1.0) # Simplified noise
)
}Methods: environmental data simulation
# calculate seasonal temp metrics
seasonal_data <- sites %>%
mutate(
temp_data = map(1:n(), ~simulate_temps()),
metrics = map(temp_data, ~{
.x %>%
mutate(season = case_when(
month(date) %in% 3:5 ~ "Spring",
month(date) %in% 6:8 ~ "Summer",
month(date) %in% 9:11 ~ "Fall",
TRUE ~ "Winter"
)) %>%
group_by(season) %>%
summarise(
avg_temp = mean(temp),
max_temp = max(temp),
min_temp = min(temp),
temp_range = max_temp - min_temp,
warm_anomaly_days = sum(temp > quantile(temp, 0.9)),
cold_anomaly_days = sum(temp < quantile(temp, 0.1)),
cumulative_gdd = sum(pmax(temp - 5, 0))
)
})
) %>%
unnest(metrics) %>%
select(-temp_data)Preliminary results: alignment
- .sam (sequence alignment map) files stores information for sequences mapped to the reference genome.
- contain a header with reference sequence name and length:
Preliminary results: alignment
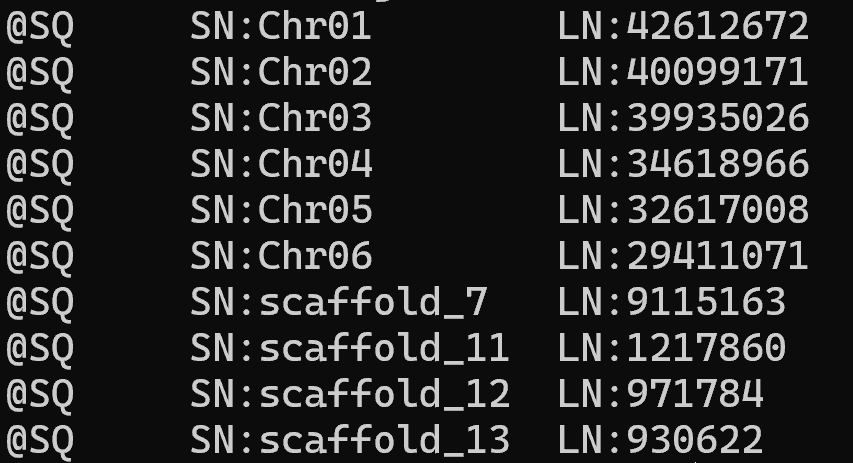
Preliminary results: alignment
- .bam (binary alignment map) files are the compressed binary version of .sam files stores information for sequences mapped to the reference genome.
- their reduced file size improves runtime for gstacks.
Preliminary results: genotyping
- gstacks is currently running! …
Preliminary results: genotyping
Preliminary results: simulating environmental data
Next four weeks
Filtering
- gstacks will produce .vcf files (variant calls and genotypes)
- remove low-quality variants and genotypes
Calculate population allele frequencies
…continued
Process and summarize environmental data at the population level (awaiting access, but I have simulated data if need be)
Build GEA analyses! Likely 2 separate analyses for cross-validation
Build workflow for identifing candidate loci