---
author: Sam White
toc-title: Contents
toc-depth: 5
toc-location: left
date: 2018-08-07 17:44:23+00:00
layout: post
slug: genome-annotation-olympia-oyster-genome-using-wq-maker-instance-on-jetstream
title: Genome Annotation - Olympia oyster genome using WQ-MAKER Instance on Jetstream
categories:
- 2018
- Olympia Oyster Genome Sequencing
tags:
- annotation
- Atmosphere
- Cyverse
- genome
- JetStream
- maker
- olympia oyster
- Ostrea lurida
- wq-maker
- Xsede
---
Yesterday, our [Xsede Startup Application (Google Doc)](https://docs.google.com/document/d/1v4ukb4M3ZY73KaBsYjcmF35pAE2pEGH9AevdXAxCONI/edit?usp=sharing) got approval for 100,000 Service Units (SUs) and 1TB of disk space on Xsede/Atmosphere/Jetstream (or, whatever it's actually called!). The approval happened within an hour of submitting the application!
Here's a copy of the approval notice:
Dear Dr. Roberts:
Your recently submitted an XSEDE Startup request has been reviewed and approved.
PI: Steven Roberts, University of Washington
Request: Annotation of Olympia oyster (Ostrea lurida) and Pacific geoduck (Panopea generosa) genomes using WQ_MAKER on Jetstream cloud.
Request Number: MCB180124 (New)
Start Date: N/A
End Date: 2019-08-05
Awarded Resources: IU/TACC (Jetstream): 100,000.0 SUs
IU/TACC Storage (Jetstream Storage): 1,000.0 GB
Allocations Admin Comments:
The estimated value of these awarded resources is $14,890.00. The allocation of these resources represents a considerable investment by the NSF in advanced computing infrastructure for U.S. The dollar value of your allocation is estimated from the NSF awards supporting the allocated resources.
If XSEDE Extended Collaborative Support (ECSS) assistance was recommended by the review panel, you will be contacted by the ECSS team within the next two weeks to begin discussing this collaboration.
By default the PI and all co-PIs will be added to the resources awarded. If this is an award on a renewal request, current users will have their account end dates modified to reflect the new end date of this award. PIs, co-PIs, or Allocation Managers can add users to or remove users from resources on this project by logging into the portal (https://portal.xsede.org) and using the 'Add/Remove User' form.
Share the impact of XSEDE! In exchange for access to the XSEDE ecosystem, we ask that all users let us know what XSEDE has helped you achieve:
>
>
> * For all publications, please acknowledge use of XSEDE and allocated resources by citing the XSEDE paper (https://www.xsede.org/how-to-acknowledge-xsede) and also add your publications to your user profile.
>
> * Tell us about your achievements (https://www.xsede.org/group/xup/science-achievements).
>
> * Help us improve our reporting by keeping your XSEDE user profile up to date and completing the demographic fields (https://portal.xsede.org/group/xup/profile).
>
For question regarding this decision, please contact help@xsede.org.
Best regards,
XSEDE Resource Allocations Service
===========================
REVIEWER COMMENTS
===========================
Review #0 - Excellent
Assessment and Summary:
Maker is a well-known and fitting workflow for Jetstream. This should be a good use of resources.
Appropriateness of Methodology:
_After_ each user on the allocation has logged in, the PI will need to open a ticket via help@xsede.org and request the quota be set per the allocation to 1TB. Please provide the XSEDE portal ID of each user.
Appropriateness of Computational Research Plan:
We request that any publications stemming from work done using Jetstream cite us - https://jetstream-cloud.org/research/citing-jetstream.php
Efficient Use of Resources:
* * *
We had a tremendous amount of help from [Upendra Devisetty at CyVerse](https://www.cyverse.org/upendra-devisetty) in getting the [Xsede](https://portal.xsede.org) Startup Application written, as well as running WQ-MAKER on Xsede/Atmosphere/Jetstream (or, whatever it's called!).
* * *
Now, on to how I got the run going...
I initiated the Olympia oyster genome annotation using a [WQ-MAKER instance (MAKER 2.31.9 with CCTools v3.1) on Jetstream](https://use.jetstream-cloud.org/application/images/492):

* * *
I followed the excellent step-by-step directions here:
* [MAKER 2.31.9 Jetstream Tutorial](https://wiki.cyverse.org/wiki/display/TUT/MAKER+2.31.9+with+CCTOOLS+Jetstream+Tutorial)
The "MASTER" machine was a "m1.xlarge" machine (i.e. CPU: 24, Mem: 60 GB, Disk: 60 GB).
I attached a 1TB volume to the MASTER machine.
I set up the run using 21 "WORKER" machines. Twenty WORKERS were "m1.large" machines (i.e. CPU: 10, Mem: 30 GB, Disk: 60 GB). The remaining WORKER was set at "m1.xlarge" (i.e. CPU: 24, Mem: 60 GB, Disk: 60 GB) to use up the rest of our allocated memory.

* * *
MASTER machine was initialized with the following command:
nohup wq_maker -contigs-per-split 1 -cores 1 -memory 2048 -disk 4096 -N wq_maker_oly${USER} -d all -o master.dbg -debug_size_limit=0 -stats test_out_stats.txt > log_file.txt 2>&1 &
Each WORKER was started with the following command:
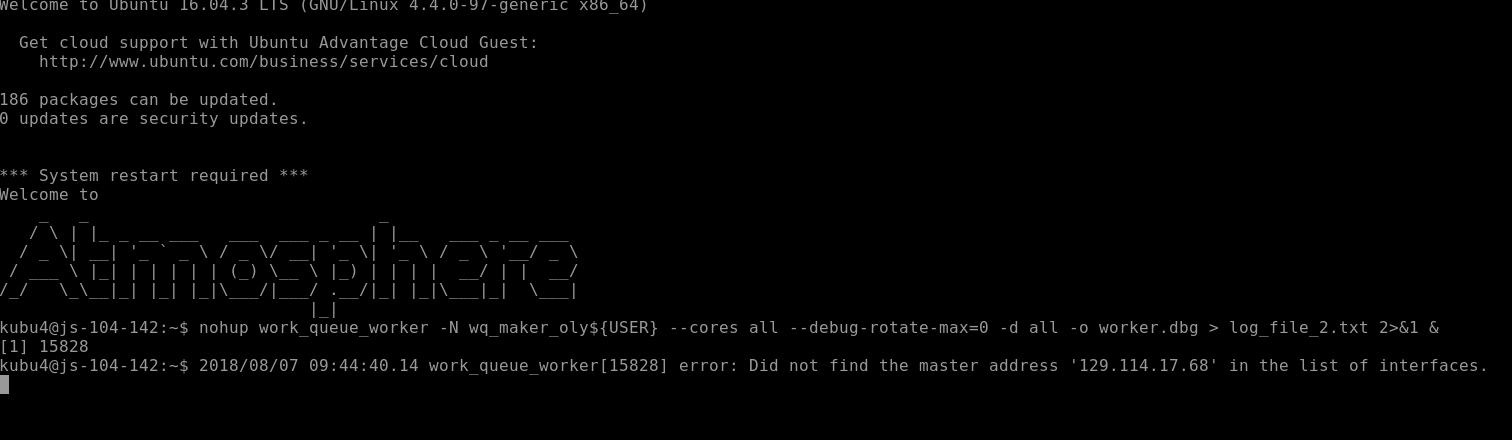
nohup work_queue_worker -N wq_maker_oly${USER} --cores all --debug-rotate-max=0 -d all -o worker.dbg > log_file_2.txt 2>&1 &
When starting each WORKER, an error message was generated, but this doesn't seem to have any impact on the ability of the program to run:
* * *
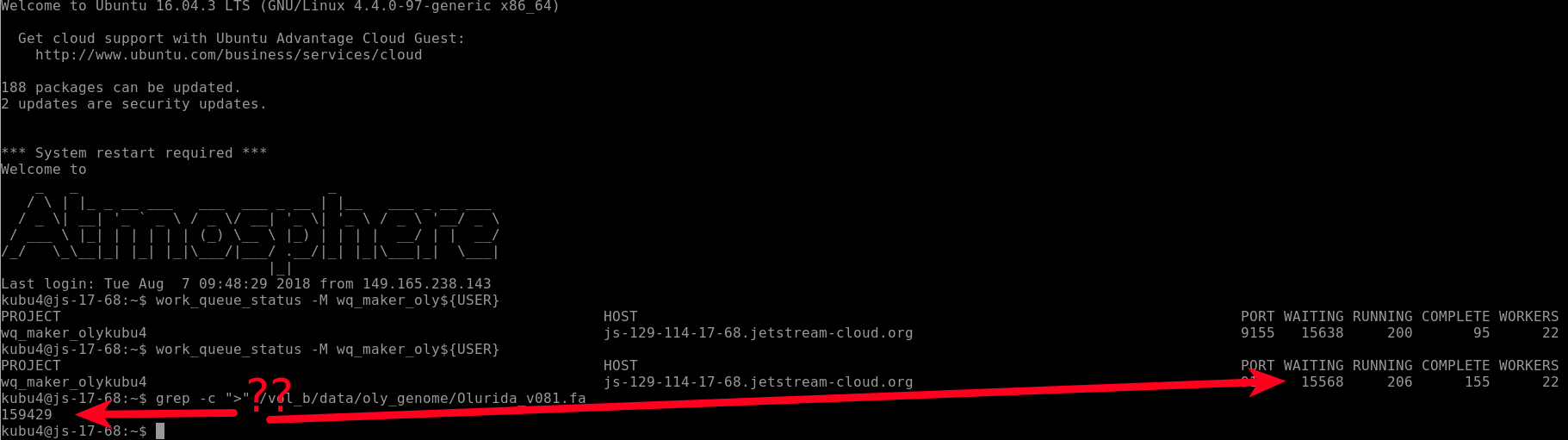
I checked on the status of the run and you can see that there are 22 WORKERS and 15,568 files "WAITING". BUT, there are 159,429 contigs in our genome FastA!
Why don't these match??!!
This is because WQ-MAKER splits the genome FastA into smaller FastA files containg only 10 sequences each. This is why we see 10-fold fewer files being processed than sequences in our genome file.
* * *
Here is the rest of the nitty gritty details:
##### Genome file:
* [Olurida_v081.fa](https://owl.fish.washington.edu/halfshell/genomic-databank/Olurida_v081.fa)
* See our [Genomic Resources GtiHub wiki for deets](https://github.com/RobertsLab/resources/wiki/Genomic-Resources#ostrea-lurida)
##### Transcriptome file:
* [Olurida_transcriptome_v3.fasta](https://eagle.fish.washington.edu/cnidarian/Olurida_transcriptome_v3.fasta)
##### Proteome files (NCBI):
The two FastA files below were concatenated into a single FastA file (`gigas_virginica_ncbi_proteomes.fasta`) for use in WQ-MAKER.
* _Crassostrea gigas_
* [GCA_000297895.1_oyster_v9_protein.faa.gz](ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/297/895/GCA_000297895.1_oyster_v9/GCA_000297895.1_oyster_v9_protein.faa.gz)
* _Crassostrea virginica_
* [GCF_002022765.2_C_virginica-3.0_protein.faa.gz](ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/002/022/765/GCF_002022765.2_C_virginica-3.0/GCF_002022765.2_C_virginica-3.0_protein.faa.gz)
##### Maker options control file (maker_opts.ctl):
This is a bit difficult to read in this notebook; copy and paste in text editor for easier viewing.
NOTE: Paths to files (e.g. genome FastA) have to be _relative_ paths; cannot be absolute paths!
#-----Genome (these are always required)
genome=../data/oly_genome/Olurida_v081.fa #genome sequence (fasta file or fasta embeded in GFF3 file)
organism_type=eukaryotic #eukaryotic or prokaryotic. Default is eukaryotic
#-----Re-annotation Using MAKER Derived GFF3
maker_gff= #MAKER derived GFF3 file
est_pass=0 #use ESTs in maker_gff: 1 = yes, 0 = no
altest_pass=0 #use alternate organism ESTs in maker_gff: 1 = yes, 0 = no
protein_pass=0 #use protein alignments in maker_gff: 1 = yes, 0 = no
rm_pass=0 #use repeats in maker_gff: 1 = yes, 0 = no
model_pass=0 #use gene models in maker_gff: 1 = yes, 0 = no
pred_pass=0 #use ab-initio predictions in maker_gff: 1 = yes, 0 = no
other_pass=0 #passthrough anyything else in maker_gff: 1 = yes, 0 = no
#-----EST Evidence (for best results provide a file for at least one)
est=../data/oly_transcriptome/Olurida_transcriptome_v3.fasta #set of ESTs or assembled mRNA-seq in fasta format
altest= #EST/cDNA sequence file in fasta format from an alternate organism
est_gff= #aligned ESTs or mRNA-seq from an external GFF3 file
altest_gff= #aligned ESTs from a closly relate species in GFF3 format
#-----Protein Homology Evidence (for best results provide a file for at least one)
protein=../data/gigas_virginica_ncbi_proteomes.fasta #protein sequence file in fasta format (i.e. from mutiple oransisms)
protein_gff= #aligned protein homology evidence from an external GFF3 file
#-----Repeat Masking (leave values blank to skip repeat masking)
model_org=all #select a model organism for RepBase masking in RepeatMasker
rmlib= #provide an organism specific repeat library in fasta format for RepeatMasker
repeat_protein= #provide a fasta file of transposable element proteins for RepeatRunner
rm_gff= #pre-identified repeat elements from an external GFF3 file
prok_rm=0 #forces MAKER to repeatmask prokaryotes (no reason to change this), 1 = yes, 0 = no
softmask=1 #use soft-masking rather than hard-masking in BLAST (i.e. seg and dust filtering)
#-----Gene Prediction
snaphmm= #SNAP HMM file
gmhmm= #GeneMark HMM file
augustus_species= #Augustus gene prediction species model
fgenesh_par_file= #FGENESH parameter file
pred_gff= #ab-initio predictions from an external GFF3 file
model_gff= #annotated gene models from an external GFF3 file (annotation pass-through)
est2genome=0 #infer gene predictions directly from ESTs, 1 = yes, 0 = no
protein2genome=0 #infer predictions from protein homology, 1 = yes, 0 = no
trna=0 #find tRNAs with tRNAscan, 1 = yes, 0 = no
snoscan_rrna= #rRNA file to have Snoscan find snoRNAs
unmask=0 #also run ab-initio prediction programs on unmasked sequence, 1 = yes, 0 = no
#-----Other Annotation Feature Types (features MAKER doesn't recognize)
other_gff= #extra features to pass-through to final MAKER generated GFF3 file
#-----External Application Behavior Options
alt_peptide=C #amino acid used to replace non-standard amino acids in BLAST databases
cpus=1 #max number of cpus to use in BLAST and RepeatMasker (not for MPI, leave 1 when using MPI)
#-----MAKER Behavior Options
max_dna_len=100000 #length for dividing up contigs into chunks (increases/decreases memory usage)
min_contig=1 #skip genome contigs below this length (under 10kb are often useless)
pred_flank=200 #flank for extending evidence clusters sent to gene predictors
pred_stats=0 #report AED and QI statistics for all predictions as well as models
AED_threshold=1 #Maximum Annotation Edit Distance allowed (bound by 0 and 1)
min_protein=0 #require at least this many amino acids in predicted proteins
alt_splice=0 #Take extra steps to try and find alternative splicing, 1 = yes, 0 = no
always_complete=0 #extra steps to force start and stop codons, 1 = yes, 0 = no
map_forward=0 #map names and attributes forward from old GFF3 genes, 1 = yes, 0 = no
keep_preds=0 #Concordance threshold to add unsupported gene prediction (bound by 0 and 1)
split_hit=10000 #length for the splitting of hits (expected max intron size for evidence alignments)
single_exon=0 #consider single exon EST evidence when generating annotations, 1 = yes, 0 = no
single_length=250 #min length required for single exon ESTs if 'single_exon is enabled'
correct_est_fusion=0 #limits use of ESTs in annotation to avoid fusion genes
tries=2 #number of times to try a contig if there is a failure for some reason
clean_try=0 #remove all data from previous run before retrying, 1 = yes, 0 = no
clean_up=0 #removes theVoid directory with individual analysis files, 1 = yes, 0 = no
TMP= #specify a directory other than the system default temporary directory for temporary files
After the run finishes, it will have produced a corresponding GFF file for each scaffold. This is unwieldly, so the GFFs will be merged using the following code:
$ gff3_merge -n -d Olurida_v081.maker.output/Olurida_v081_master_datastore_index.log
All WORKERS were running as of 07:45 today. As of this posting (~3hrs later), WQ-MAKER had already processed ~45% of the files! Annotation will be finished by the end of today!!! Crazy!