---
author: Sam White
toc-title: Contents
toc-depth: 5
toc-location: left
date: 2018-07-11 20:11:05+00:00
layout: post
slug: mox-olympia-oyster-genome-annotation-progress-using-maker-2-31-10
title: Mox - Olympia oyster genome annotation progress (using Maker 2.31.10)
categories:
- 2018
- Olympia Oyster Genome Sequencing
tags:
- maker
- mox
- olympia oyster
- Ostrea lurida
---
#### TL;DR - It appears to be continuing where it left off!
I decided to spend some time to figure out what was actually happening, as it's clear that the annotation process is going to need some additional time to run and may span an additional monthly maintenance shutdown.
This is great, because, otherwise, this will take an eternity to actually complete (particularly because we'd have to move the job to run on one of our lab's computers - which pale in comparison to the specs of our Mox nodes).
However, it's a bit shocking that this is taking this long, even on a Mox node!
I started annotating the Olympia oyster genome on 20180529. Since then, the job has been interrupted twice by monthly Mox maintenance (which happens on the 2nd Tuesday of each month). Additionally, when this happens, the SLURM output file is overwritten, making it difficult to assess whether or not Maker continues where it left off or if it's starting over from scratch.
Anyway, here's how I deduced that the program is continuing where it left off.
1. I figured out that it produces a generic feature format (GFF) file for each contig.
2. Decided to search for the first contig GFF and look at it's last modified date. This would tell me if it was newly generated (i.e. on the date that the job was restarted after the maintenance shutdown) or if it was old. Additionally, if there were more than one of these files, then I'd also know that Maker was just starting at the beginning and writing data to a different location.
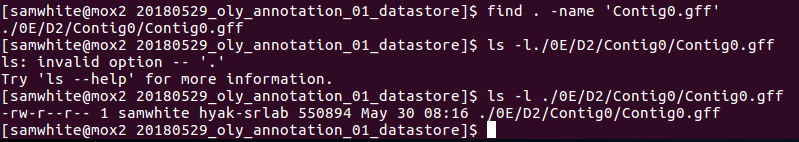
This shows:
1. Only one copy of Contig0.gff exists.
2. Last modified date is 20180530.
3. Check the slurm output file for info.
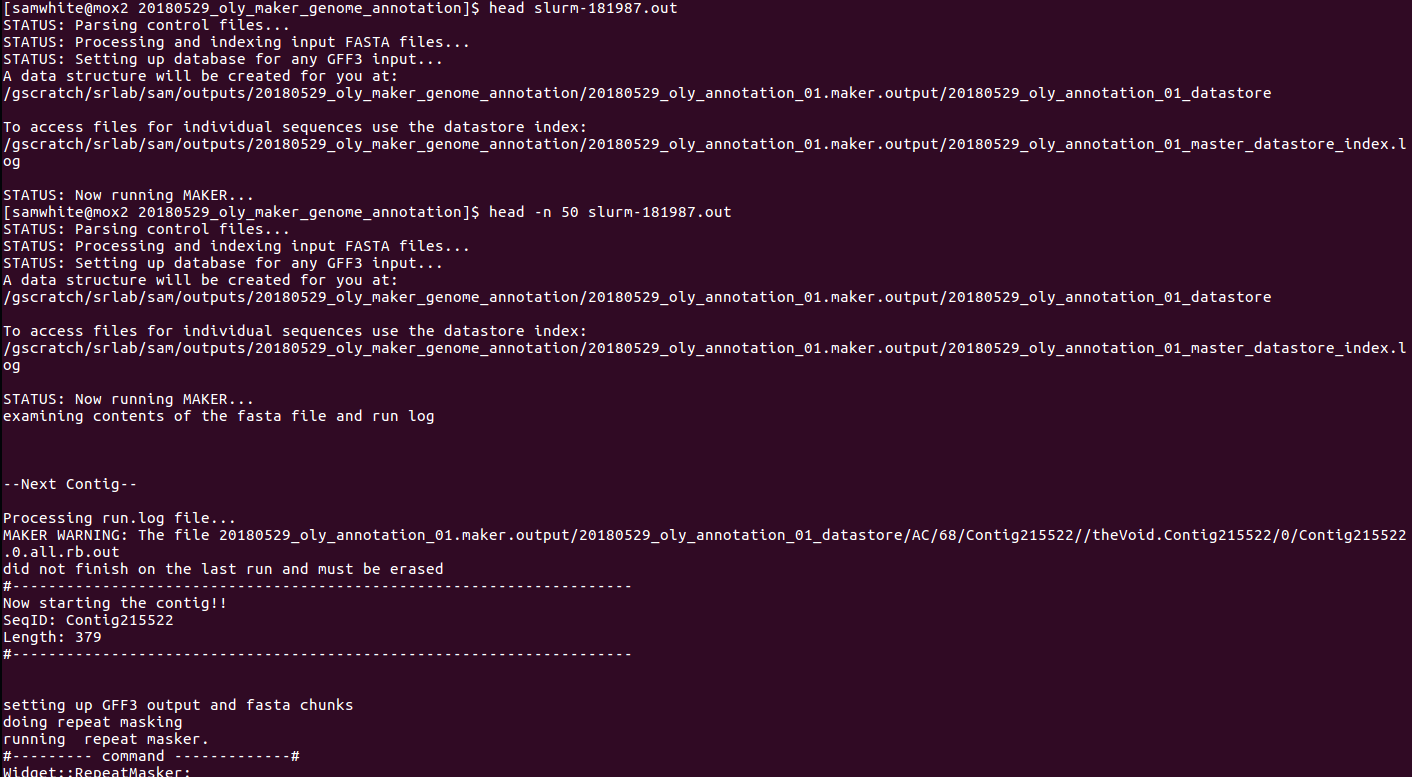
This reveals this important piece of info:
MAKER WARNING: The file 20180529_oly_annotation_01.maker.output/20180529_oly_annotation_01_datastore/AC/68/Contig215522//theVoid.Contig215522/0/Contig215522.0.all.rb.out
did not finish on the last run
All of these taken together lead me to confidently conclude that Maker is _not_ restarting from the beginning and is, indeed, continuing where it left off. WHEW!
Just for kicks, I also ran a count of GFF files to see where this stands so far:
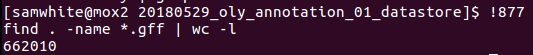
Wow! 622,010 GFFs!!!
Finally, for posterity, here's the SLURM script I used to submit this job, back in May! I'll have all of the corresponding genome files, proteome files, transcriptome files, etc. on one of our servers once the job completes.
#!/bin/bash
## Job Name
#SBATCH --job-name=20180529_oly_maker_genome_annotation
## Allocation Definition
#SBATCH --account=srlab
#SBATCH --partition=srlab
## Resources
## Nodes
#SBATCH --nodes=1
## Walltime (days-hours:minutes:seconds format)
#SBATCH --time=30-00:00:00
## Memory per node
#SBATCH --mem=500G
##turn on e-mail notification
#SBATCH --mail-type=ALL
#SBATCH --mail-user=samwhite@uw.edu
## Specify the working directory for this job
#SBATCH --workdir=/gscratch/srlab/sam/outputs/20180529_oly_maker_genome_annotation
## Establish variables for more readable code
### Path to Maker executable
maker=/gscratch/srlab/programs/maker-2.31.10/bin/maker
### Path to Olympia oyster genome FastA file
oly_genome=/gscratch/srlab/sam/data/O_lurida/oly_genome_assemblies/jelly.out.fasta
### Path to Olympia oyster transcriptome FastA file
oly_transcriptome=/gscratch/srlab/sam/data/O_lurida/oly_transcriptome_assemblies/Olurida_transcriptome_v3.fasta
### Path to Crassotrea gigas NCBI protein FastA
gigas_proteome=/gscratch/srlab/sam/data/C_gigas/gigas_ncbi_protein/GCA_000297895.1_oyster_v9_protein.faa
### Path to Crassostrea virginica NCBI protein FastA
virginica_proteome=/gscratch/srlab/sam/data/C_virginica/virginica_ncbi_protein/GCF_002022765.2_C_virginica-3.0_protein.faa
## Create Maker control files needed for running Maker
$maker -CTL
## Store path to options control file
maker_opts_file=./maker_opts.ctl
## Create combined proteome FastA file
touch gigas_virginica_ncbi_proteomes.fasta
cat "$gigas_proteome" >> gigas_virginica_ncbi_proteomes.fasta
cat "$virginica_proteome" >> gigas_virginica_ncbi_proteomes.fasta
## Edit options file
### Set paths to O.lurida genome and transcriptome.
### Set path to combined C. gigas and C.virginica proteomes.
## The use of the % symbol sets the delimiter sed uses for arguments.
## Normally, the delimiter that most examples use is a slash "/".
## But, we need to expand the variables into a full path with slashes, which screws up sed.
## Thus, the use of % symbol instead (it could be any character that is NOT present in the expanded variable; doesn't have to be "%").
sed -i "/^genome=/ s% %$oly_genome %" "$maker_opts_file"
sed -i "/^est=/ s% %$oly_transcriptome %" "$maker_opts_file"
sed -i "/^protein=/ s% %$gigas_virginica_ncbi_proteomes %" "$maker_opts_file"
## Run Maker
### Set basename of files and specify number of CPUs to use
$maker \
-base 20180529_oly_annotation_01 \
-cpus 24