{
"cells": [
{
"cell_type": "markdown",
"metadata": {},
"source": [
"# Data-Independent Mass Spectrometry: From RAW files to Analyses\n",
"\n",
"In this notebook, I'll outline the workflow necessary to take my *Crassostrea gigas* Data-Independent Mass Spectrometry (DIA) data from `RAW` files off of the mass spectrometer to a `.csv` usable in R Studio. DIA allows us to identify all peptides in each oyster sample without having any prior knowledge of what we may find.\n",
"\n",
"*Note: This pipeline relies heavily on Windows-based programs*"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Step 1: Collect Materials"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"**Software Dependencies**:\n",
"\n",
"- [Skyline daily](https://skyline.ms/project/home/software/Skyline/daily/register-form/begin.view?)\n",
" - This version of Skyline will udpates as the software is modified. You need permission to download it. The version of Skyline daily used in this notebook is Skyline-daily (64-bit) 3.7.1.11446 (as of 2017-10-11). \n",
"- [MSConvert]()\n",
" - Special version of MSConvert modified by Austin in Genome Sciences on 2017-04-18.\n",
"- [R](https://www.r-project.org) and [R Studio](https://www.rstudio.com)\n",
" - R version 3.4.0 (2017-04-21) -- \"You Stupid Darkness\"\n",
" - R Studio Version 1.0.143\n",
" \n",
"**Specific files (in order used)**:\n",
"\n",
"- [config file for MSConvert](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_MSConvert_20170412/config_fix_20170413.txt)\n",
" - This file contains all of the information MSConvert needs to demultiplex RAW files. It includes an arugment specific to the isolation scheme used by the mass spectrometer to collect data.\n",
"- [.blib file from `pecanpie`](http://owl.fish.washington.edu/spartina/DNR_Skyline_20170524/2017-05-23-oyster-desearleinated.blib)\n",
" - A .blib file is a library of all peptides you want to identify in your samples. For this experiment, the .blib contains the entire *C. gigas* proteome. This is the same .blib file used in my DIA analyses. This file was created by Emma Timmins-Schiffman and her team in `brecan`, a version of `pecanpie` produced by Brian Searle in Genome Sciences. Jarrett Egertson wrote a script to get rid of incompatibilities generated by `brecan`. The final compatible document is found above. For more information regarding .blib creation, see this [lab notebook entry](https://yaaminiv.github.io/Skyline-Attempt-3/).\n",
"- [Undigested **FASTA** background proteome, including QC protein sequence](http://owl.fish.washington.edu/spartina/DNR_Skyline_20170427/Combined-gigas-QC.fasta)\n",
"- [RAW files](http://owl.fish.washington.edu/spartina/January_2017_DNR_Raw_Data/Oyster_raw_files/)\n",
"- [Key for samples](http://owl.fish.washington.edu/spartina/January_2017_DNR_Raw_Data/2017_January_23.csv)"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Step 2: Demultiplex RAW data in MSConvert\n",
"\n",
"DIA mass spectrometry uses overlapping mass-charge (m/z) windows to capture all peptides in a sample. To discern which peptides and transitions were found in each window, RAW files are demultiplexed. This allows us to parse out peptide abundance data from each individual window. Demultiplexing requires use of the MSConvert command line interface."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 2a: Install the MSConvert CLI\n",
"\n",
"Unzip the file in Step 1 in your desired working directory. This should be the same directory with the RAW files to convert."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 2b: Convert RAW files\n",
"\n",
"Use the following code to demultiplex all RAW files and convert them to .mzML files:\n",
"\n",
"`msconvert.exe -c config_fix_20170413.txt *.raw`"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Step 3: Prepare Peptide Database"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 3a. Merge reference proteome with Quality Standards.\n",
"\n",
"Essentially concatenate the fasta files. \n",
"\n",
"[C. gigas proteome](http://owl.fish.washington.edu/halfshell/bu-git-repos/nb-2017/C_gigas/data/Cg_Gigaton_proteins.fa). \n",
"A tabular version of the QC peptide list for the January 2017 Lumos run can be found [here](http://owl.fish.washington.edu/generosa/Generosa_DNR/Pierce_PRTC.tabular)."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 3b. Run in silico tryptic digestion of reference proteome with quality standards.\n",
"\n",
"This is done with [Protein Digestion Simulator](https://omics.pnl.gov/software/protein-digestion-simulator), a Windows program.\n",
"\n",
"Settings for the in silico tryptic digest, as discussed on [Issue #483](https://github.com/sr320/LabDocs/issues/483). Each screenshot refers to a different tab in the program window. The screenshots are sequential (Tab #1-Tab #4).\n",
"\n",
"Ensure the output, a digested reference proteome, is in a .tabular format.\n",
"\n",
"\n",
"\n",
"*Note: \"Delimited Input File Options\" depends on the format of your fasta file. This one only has protein ID and sequence.*\n",
"\n",
"\n",
"\n",
"\n",
"\n",
"*Note: Use default settings*\n",
"\n",
"\n",
"\n",
"*Note: Use default settings*\n",
"\n",
"To execute digest, on select \"Parse and Digest\" on Tab #2."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 3c. Modify .tabular digested proteome\n",
"\n",
"The digested proteome will provide information unnecessary for PECAN.\n",
"\n",
"\n",
"\n",
"The only columns needed for PECAN are the first two: \"Protein_Name\" and \"Sequence\". These columns can be cut out using `awk` or in [Galaxy](usegalaxy.org). The final .tabular digested proteome should look like this:\n",
"\n",
"\n",
"\n",
"Example\n",
"On Windows using Git Bash.\n",
"```\n",
"\n",
"srlab@swan MINGW64 ~\n",
"$ cd Desktop/\n",
"\n",
"srlab@swan MINGW64 ~/Desktop\n",
"$ cd grace/\n",
"\n",
"srlab@swan MINGW64 ~/Desktop/grace\n",
"$ head Cg_Giga_cont_prtc_AA_digested_Mass400to6000.txt\n",
"Protein_Name Sequence Unique_ID Monoisotopic_Mass Predicte\n",
"d_NET Tryptic_Name\n",
"CHOYP_043R.5.5|m.64252 SPSEDPDAPIENILQTNSVYKPK 1 2541.2598016 0.3655t2\n",
".1\n",
"CHOYP_043R.5.5|m.64252 SPSEDPDAPIENILQTNSVYKPKK 2 2669.35475980.34\n",
"14 t2.2\n",
"CHOYP_043R.5.5|m.64252 SPSEDPDAPIENILQTNSVYKPKKEPTYDENVVVK 3 3942.973\n",
"762 0.3449 t2.3\n",
"CHOYP_043R.5.5|m.64252 SPSEDPDAPIENILQTNSVYKPKKEPTYDENVVVKIISQDTPTILR 45180.67\n",
"6764 0.5144 t2.4\n",
"CHOYP_043R.5.5|m.64252 KEPTYDENVVVK 5 1419.7245246 0.2186 t3.2\n",
"CHOYP_043R.5.5|m.64252 KEPTYDENVVVKIISQDTPTILR 6 2657.4275266 0.4593t3\n",
".3\n",
"CHOYP_043R.5.5|m.64252 KEPTYDENVVVKIISQDTPTILRVSFTVNR 7 3460.85649280.56\n",
"58 t3.4\n",
"CHOYP_043R.5.5|m.64252 EPTYDENVVVK 8 1291.6295664 0.2301 t4.1\n",
"CHOYP_043R.5.5|m.64252 EPTYDENVVVKIISQDTPTILR 9 2529.3325684 0.4402t4\n",
".2\n",
"\n",
"srlab@swan MINGW64 ~/Desktop/grace\n",
"$ awk '{print $1,$2}' Cg_Giga_cont_prtc_AA_digested_Mass400to6000.txt | head\n",
"Protein_Name Sequence\n",
"CHOYP_043R.5.5|m.64252 SPSEDPDAPIENILQTNSVYKPK\n",
"CHOYP_043R.5.5|m.64252 SPSEDPDAPIENILQTNSVYKPKK\n",
"CHOYP_043R.5.5|m.64252 SPSEDPDAPIENILQTNSVYKPKKEPTYDENVVVK\n",
"CHOYP_043R.5.5|m.64252 SPSEDPDAPIENILQTNSVYKPKKEPTYDENVVVKIISQDTPTILR\n",
"CHOYP_043R.5.5|m.64252 KEPTYDENVVVK\n",
"CHOYP_043R.5.5|m.64252 KEPTYDENVVVKIISQDTPTILR\n",
"CHOYP_043R.5.5|m.64252 KEPTYDENVVVKIISQDTPTILRVSFTVNR\n",
"CHOYP_043R.5.5|m.64252 EPTYDENVVVK\n",
"CHOYP_043R.5.5|m.64252 EPTYDENVVVKIISQDTPTILR\n",
"\n",
"srlab@swan MINGW64 ~/Desktop/grace\n",
"$ awk '{print $1,$2}' Cg_Giga_cont_prtc_AA_digested_Mass400to6000.txt \\\n",
"> > Cg_Giga_cont_prtc_AA_M400-6000-2c.txt\n",
"```"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Step 4. Run PECAN\n",
"\n",
"[PECAN](https://bitbucket.org/maccosslab/pecan/overview) correlates your acquired peptide spectra to a database of known sequences and creates a library of proteins and peptides that you detected in your experiment. PECAN requires several inputs, each of which must be prepared before running PECAN in the command line.\n",
"\n",
"### Step 4a. Create two text files that indicate path for 1) the reference peptide list (in silico protein digestion) and 2) spectra data (mzML files)\n",
"\n",
"The background proteome should be a .txt list with protein names and sequences. It can be the same file as the one generated by the in silico protein digestion step above.\n",
"\n",
"[example peptide path file](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_PECAN_Run_3_20170308/2017-03-08-background-peptides-path-list.txt)\n",
"\n",
"Similar to the peptide path file, this should be a .txt file with the path for each of your `.mzML` files you want to analyze.\n",
"\n",
"[example .mzmL path file](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_PECAN_Run_3_20170308/2017-03-08-mzML-file-path-list.txt)\n",
"\n",
"### Step 4b. Generate isolation scheme file\n",
"\n",
"The isolation scheme is a list of DIA precursor windows with m/z ratios used to analyze samples. The isolation schemes are experiment-dependent, so you need to look at your mass spectrometry method file to figure it out. The final isolation scheme should be formatted as a .csv file. The m/z ratios should be paired.\n",
"\n",
"[example isolation scheme](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/2018-02-28-PECAN/PECAN-inputs/2017-03-03-isolation-windows.csv)\n",
"\n",
"### Step 4c. Add Background Proteome to Pecan\n",
"\n",
"Pecan needs to be compiled with the background proteome. To do this: \n",
"\n",
"1. Copy background proteome to `/home/shared/pecan/PECAN/PecanUtil/` \n",
"\n",
"2. Add the background proteome to the config file via `gedit /home/shared/pecan/PECAN/PecanUtil/config`. It will be added to the end of the file and follow the format `speciesname = file name` ex: `geoduck = geoduck-background-proteome.csv` \n",
"\n",
"3. Recompile Pecan via `python /home/shared/pecan/setup.py install` \n",
"\n",
"### Step 4d. Create a peptide spectral library from your data\n",
"\n",
"This will be done using the `pecanpie` command and the code below:\n",
"\n",
"```\n",
"pecanpie -o [directory to create] \\ # This is the directory you will navigate to afterwards to run a search.\n",
"-b [digested background proteome] \\\n",
"-n [blib file name] \\ # The name of the file you will use in Skyline. No need to include a .blib extension.\n",
"-s [species] \\ # The species may need to be configured beforehand every time this command is run.\n",
"--isolationSchemeType BOARDER \\ \n",
"--pecanMemRequest [GB estimate] \\ # PECAN will ask you to input a GB estimate of the memory you need. If your estimate is too low, the program will suggest a higher memory for you to use.\n",
"[filename for mzML file path list] \\ \n",
"[filename for peptide file path] \\ \n",
"[filename for isolation scheme file name] \\\n",
"--fido --jointPercolator\n",
"```\n",
"\n",
"This step will be fast because it is just setting up the real PECAN run. Navigate to your newly created directory (specified by the `-o` argument and run the actual search: \n",
"\n",
"```\n",
"cd [directory specified by the -o argument for pecanpie] \\\n",
"./run_search.sh\n",
"```\n",
"\n",
"The search takes a while to run, especially if you are looking for all possible peptides in your proteome. You can check the status of your job with the following code:\n",
"\n",
"```\n",
"qstat -f\n",
"```\n",
"\n",
"### Step 4e. Confirm creation of .blib file\n",
"\n",
"When `pecanpie` is done running, navigate to the `pecan2blib` folder within your output directory. Inside that folder, you'll find the .blib file, the name of which is specified by `pecanpie -n`.\n",
"\n",
"The .blib file you create will be used in the Skyline workflow."
]
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"## Step 3: Create a new Skyline Document"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 3a: Import the Spectral Library\n",
"\n",
"1. Open a new Skyline file (\"Blank Document\")\n",
"2. Under Settings >> Peptide Settings >> Library, click \"Edit List.\" The *C. gigas* .blib should already be on the list (2017-05-23-oyster-desearlinated).\n",
"3. If the .blib is not already on the list, click \"Add\"\n",
" - Name the library and select the .blib file\n",
" - After clicking \"OK,\" select the correct library from the list\n",
"4. Under pick peptides matching, select \"Library\"\n",
"\n",
" "
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 3b: Add Background Proteome\n",
"\n",
"1. Settings >> Peptide Settings >> Digestion\n",
"2. Select \"Gigas-4-27\" under \"Background proteome\"\n",
"3. If \"Gigas-4-27\" is not an option\n",
" - Select \"Add\" under \"Background proteome\"\n",
" - Name background\n",
" - Click \"Create\" under \"Proteome file\" to choose where to save the background\n",
" - Click \"Add file\" under \"FASTA files\", and select your background proteome fasta file\n",
" - After the FASTA loads, Skyline may prompt you about deleting repeated protein sequences. Select \"OK\"\n",
" \n",
" 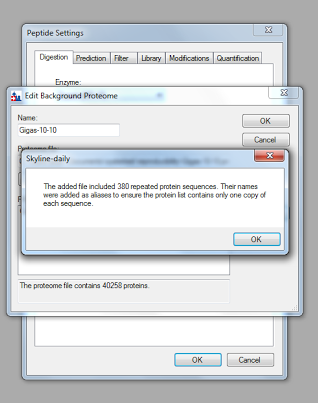 \n",
"\n",
"4. Select \"Trypsin [KR | P]\" under \"Enzyme\n",
"\n",
"
"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 3b: Add Background Proteome\n",
"\n",
"1. Settings >> Peptide Settings >> Digestion\n",
"2. Select \"Gigas-4-27\" under \"Background proteome\"\n",
"3. If \"Gigas-4-27\" is not an option\n",
" - Select \"Add\" under \"Background proteome\"\n",
" - Name background\n",
" - Click \"Create\" under \"Proteome file\" to choose where to save the background\n",
" - Click \"Add file\" under \"FASTA files\", and select your background proteome fasta file\n",
" - After the FASTA loads, Skyline may prompt you about deleting repeated protein sequences. Select \"OK\"\n",
" \n",
"  \n",
"\n",
"4. Select \"Trypsin [KR | P]\" under \"Enzyme\n",
"\n",
" "
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 3c: Adjust Peptide Settings\n",
"\n",
"1. Under the Prediction tab, make sure \"none\" is selected for retention time predictor\n",
"\n",
"
"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 3c: Adjust Peptide Settings\n",
"\n",
"1. Under the Prediction tab, make sure \"none\" is selected for retention time predictor\n",
"\n",
"  \n",
"\n",
"2. Under the Filter tab, click \"Auto-select all matching peptides\" and ensure 2 and 25 are the Min and Max lengths. Under \"Exclude N-Terminal AAs\" enter zero.\n",
"\n",
"
\n",
"\n",
"2. Under the Filter tab, click \"Auto-select all matching peptides\" and ensure 2 and 25 are the Min and Max lengths. Under \"Exclude N-Terminal AAs\" enter zero.\n",
"\n",
"  \n",
"\n",
"3. Under the Modification tab, select \"Carbamidomethyl (C)\" under structural modifications, \"heavy\" Isotope label type, and \"light\" Internal standard type.\n",
"\n",
"
\n",
"\n",
"3. Under the Modification tab, select \"Carbamidomethyl (C)\" under structural modifications, \"heavy\" Isotope label type, and \"light\" Internal standard type.\n",
"\n",
"  \n",
"\n",
"4. Use the following settings under the Quantification tab\n",
"\n",
"
\n",
"\n",
"4. Use the following settings under the Quantification tab\n",
"\n",
"  "
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 3d: Populate Analyte Tree\n",
"\n",
"1. Under File >> Import, select \"Transition List\"\n",
"2. Select the transition list (.csv) outlined in Step 1. This will populate the analyte tree only with targeted proteins and quality control (PRTC) peptides.\n",
"\n",
"Skyline will keep the proteins, peptides and transitions that match what it finds in the library provided in Step 2a."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 3e: Adjust Transition Settings\n",
"\n",
"1. **Settings >> Transition Settings >> Prediction**\n",
" - Precursor mass: Monoisotopic\n",
" - Product ion mass: Monoisotopic\n",
" - Collision energy: Thermo TSQ Vantage\n",
" - Declustering potential: None\n",
" - Optimization library: None\n",
" - Compensation voltage: None\n",
" - DO NOT select \"Use optimization values when present\"\n",
" \n",
"
"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 3d: Populate Analyte Tree\n",
"\n",
"1. Under File >> Import, select \"Transition List\"\n",
"2. Select the transition list (.csv) outlined in Step 1. This will populate the analyte tree only with targeted proteins and quality control (PRTC) peptides.\n",
"\n",
"Skyline will keep the proteins, peptides and transitions that match what it finds in the library provided in Step 2a."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 3e: Adjust Transition Settings\n",
"\n",
"1. **Settings >> Transition Settings >> Prediction**\n",
" - Precursor mass: Monoisotopic\n",
" - Product ion mass: Monoisotopic\n",
" - Collision energy: Thermo TSQ Vantage\n",
" - Declustering potential: None\n",
" - Optimization library: None\n",
" - Compensation voltage: None\n",
" - DO NOT select \"Use optimization values when present\"\n",
" \n",
"  \n",
"\n",
"2. **Settings >> Transition Settings >> Filter >> Peptides**\n",
" - Precursor charges: 2, 3\n",
" - Ion charges: 1, 2\n",
" - Ion types: y\n",
" - Product ion selection\n",
" - From: ion 2\n",
" - To: last ion\n",
" - DO NOT select any Special ions\n",
" - DO NOT specify any Precursor m/z exclusion window\n",
" - DO select \"Auto-select all matching transitions\"\n",
" \n",
"
\n",
"\n",
"2. **Settings >> Transition Settings >> Filter >> Peptides**\n",
" - Precursor charges: 2, 3\n",
" - Ion charges: 1, 2\n",
" - Ion types: y\n",
" - Product ion selection\n",
" - From: ion 2\n",
" - To: last ion\n",
" - DO NOT select any Special ions\n",
" - DO NOT specify any Precursor m/z exclusion window\n",
" - DO select \"Auto-select all matching transitions\"\n",
" \n",
"  \n",
" \n",
"3. **Transition Settings >> Library**\n",
" - Ion match tolerance: 0.5 m/z\n",
" - DO NOT select \"If a library spectrum is available, pick its most intense ions\"\n",
" \n",
"
\n",
" \n",
"3. **Transition Settings >> Library**\n",
" - Ion match tolerance: 0.5 m/z\n",
" - DO NOT select \"If a library spectrum is available, pick its most intense ions\"\n",
" \n",
"  \n",
" \n",
"4. **Transition Settings >> Instrument**\n",
" - Min m/z: 100\n",
" - Max m/z: 2000\n",
" - DO NOT select \"Dynamic min product m/z\"\n",
" - Method match tolerance m/z: 0.055 m/z\n",
" - DO NOT specify any other settings on this tab\n",
" \n",
"
\n",
" \n",
"4. **Transition Settings >> Instrument**\n",
" - Min m/z: 100\n",
" - Max m/z: 2000\n",
" - DO NOT select \"Dynamic min product m/z\"\n",
" - Method match tolerance m/z: 0.055 m/z\n",
" - DO NOT specify any other settings on this tab\n",
" \n",
"  \n",
" \n",
"5. **Transition Settings >> Full-Scan**\n",
" - Isotope peaks included: Count\n",
" - Precursor mass analyzer: Orbitrap\n",
" - Peaks: 3\n",
" - Resolving power: 60,000\n",
" - At: 400 m/z\n",
" - Isotope labeling enrichment: Default\n",
" - Acquisition method: Targeted\n",
" - Product mass analyzer: Centroided\n",
" - Isolation scheme: None\n",
" - Mass Accuracy: 20 ppm\n",
" - DO NOT select \"Use high-sensitivity extraction\"\n",
" - Select \"Use only scans within 2 minutes of MS/MS IDs\"\n",
" \n",
"
\n",
" \n",
"5. **Transition Settings >> Full-Scan**\n",
" - Isotope peaks included: Count\n",
" - Precursor mass analyzer: Orbitrap\n",
" - Peaks: 3\n",
" - Resolving power: 60,000\n",
" - At: 400 m/z\n",
" - Isotope labeling enrichment: Default\n",
" - Acquisition method: Targeted\n",
" - Product mass analyzer: Centroided\n",
" - Isolation scheme: None\n",
" - Mass Accuracy: 20 ppm\n",
" - DO NOT select \"Use high-sensitivity extraction\"\n",
" - Select \"Use only scans within 2 minutes of MS/MS IDs\"\n",
" \n",
"  "
]
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Step 5: Clean Data\n",
"\n",
"In this step, files without any data will be removed and peaks in Skyline will be verfied against predicted retention times."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 5a: Check that all QC peptides are chosen correctly"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 5b: Spot check peptides"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 5c: Export data\n",
"\n",
"To proceed with downstream analyses, data must be exported from Skyline as a .csv."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Under File > Export > Report, use the following settings to export Skyline data as a .csv. However, do not include the \"Total Ion Current Area\" option.\n",
"\n",
"\n",
"\n",
"Exported data can be found [here](http://owl.fish.washington.edu/spartina/DNR_SRM_20170728/Analyses/2017-09-12-Gigas-SRM-ReplicatesOnly-PostDilutionCurve-NoPivot-RevisedSettings-Report.csv)."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 5d: Normalize peak areas"
]
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
}
],
"metadata": {
"anaconda-cloud": {},
"kernelspec": {
"display_name": "Python [default]",
"language": "python",
"name": "python3"
},
"language_info": {
"codemirror_mode": {
"name": "ipython",
"version": 3
},
"file_extension": ".py",
"mimetype": "text/x-python",
"name": "python",
"nbconvert_exporter": "python",
"pygments_lexer": "ipython3",
"version": "3.5.2"
}
},
"nbformat": 4,
"nbformat_minor": 1
}
"
]
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Step 5: Clean Data\n",
"\n",
"In this step, files without any data will be removed and peaks in Skyline will be verfied against predicted retention times."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 5a: Check that all QC peptides are chosen correctly"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 5b: Spot check peptides"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 5c: Export data\n",
"\n",
"To proceed with downstream analyses, data must be exported from Skyline as a .csv."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Under File > Export > Report, use the following settings to export Skyline data as a .csv. However, do not include the \"Total Ion Current Area\" option.\n",
"\n",
"\n",
"\n",
"Exported data can be found [here](http://owl.fish.washington.edu/spartina/DNR_SRM_20170728/Analyses/2017-09-12-Gigas-SRM-ReplicatesOnly-PostDilutionCurve-NoPivot-RevisedSettings-Report.csv)."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 5d: Normalize peak areas"
]
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
}
],
"metadata": {
"anaconda-cloud": {},
"kernelspec": {
"display_name": "Python [default]",
"language": "python",
"name": "python3"
},
"language_info": {
"codemirror_mode": {
"name": "ipython",
"version": 3
},
"file_extension": ".py",
"mimetype": "text/x-python",
"name": "python",
"nbconvert_exporter": "python",
"pygments_lexer": "ipython3",
"version": "3.5.2"
}
},
"nbformat": 4,
"nbformat_minor": 1
}