{
"cells": [
{
"cell_type": "markdown",
"metadata": {},
"source": [
"# Selected Reaction Monitoring Data: From RAW files to Analyses\n",
"\n",
"In this notebook, I'll outline the workflow necessary to take my *Crassostrea gigas* Selected Reaction Monitoring (SRM) data from `RAW` files off of the mass spectrometer to a `.csv` usable in R Studio."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Step 1: Collect Materials"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"**Software Dependencies**:\n",
"\n",
"- [Skyline daily](https://skyline.ms/project/home/software/Skyline/daily/register-form/begin.view?)\n",
" - This version of Skyline will udpates as the software is modified. You need permission to download it. The version of Skyline daily used in this notebook is Skyline-daily (64-bit) 3.7.1.11446 (as of 2017-10-11). \n",
"- [R](https://www.r-project.org) and [R Studio](https://www.rstudio.com)\n",
" - R version 3.4.0 (2017-04-21) -- \"You Stupid Darkness\"\n",
" - R Studio Version 1.0.143\n",
" \n",
"**Specific files**:\n",
"\n",
"- [.blib file from `pecanpie`](http://owl.fish.washington.edu/spartina/DNR_Skyline_20170524/2017-05-23-oyster-desearleinated.blib?raw=true)\n",
" - A .blib file is a library of all peptides you want to identify in your samples. For this experiment, the .blib contains the entire *C. gigas* proteome. This is the same .blib file used in my DIA analyses. This file was created by Emma Timmins-Schiffman and her team in `brecan`, a version of `pecanpie` produced by Brian Searle in Genome Sciences. Jarrett Egertson wrote a script to get rid of incompatibilities generated by `brecan`. The final compatible document is found above. For more information regarding .blib creation, see this [lab notebook entry](https://yaaminiv.github.io/Skyline-Attempt-3/).\n",
"- [Undigested **FASTA** background proteome, including QC protein sequence](http://owl.fish.washington.edu/spartina/DNR_Skyline_20170427/Combined-gigas-QC.fasta)\n",
" - Right-click and select \"Save link as...\" to download\n",
"- [Transition list (.csv) for protein targets and PRTC peptides](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-08-Final-Transitions/2017-07-10-SRM-Transitions-With-PRTC.csv?raw=true)\n",
" - Right-click and select \"Save link as...\" to download\n",
"- [single zip of RAW files from mass spectrometer (171 files - < 1GB)](http://owl.fish.washington.edu/spartina/DNR_SRM_20170728/Raw-Files/Archive.zip)\n",
" - The Raw-files are split into four folders: blanks, quality control samples, oyster samples and dilution curve samples. You will only need to use the oyster samples and dilution curve samples. Be sure to unzip this archive file before you proceed.\n",
"- [Key for samples](http://owl.fish.washington.edu/spartina/DNR_SRM_20170728/Analyses/2017-07-28-SRM-Sequence-File.xlsx?raw=true)\n",
" - This sample key came straight off the mass spectrometer, with the exception of the TIC values added after samples were run.\n",
"- [Predicted retention times](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-08-Final-Transitions/2017-07-11-Predicted-SRM-Retention-Times.xlsx?raw=true)"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Step 2: Create Dilution Curve Skyline Document\n",
"\n",
"The purpose of this is to verify that the assay is detecting peptides based on their specific presence in the autosampler vial. We want to ensure that the machine only captures the oyster transitions we set out to measure. The oyster peptide detection should decrease as the concentration in the autosampler vial decreases, and vice versa."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 2a: Import the Spectral Library\n",
"\n",
"1. Open a new Skyline file (\"Blank Document\")\n",
"2. Under Settings >> Peptide Settings >> Library, click \"Edit List.\" The *C. gigas* .blib should already be on the list (2017-05-23-oyster-desearlinated).\n",
"3. If the .blib is not already on the list, click \"Add\"\n",
" - Name the library and select the .blib file\n",
" - After clicking \"OK,\" select the correct library from the list\n",
"4. Under pick peptides matching, select \"Library\"\n",
"\n",
" "
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 2b: Add Background Proteome\n",
"\n",
"1. Settings >> Peptide Settings >> Digestion\n",
"2. Select \"Gigas-4-27\" under \"Background proteome\"\n",
"3. If \"Gigas-4-27\" is not an option\n",
" - Select \"Add\" under \"Background proteome\"\n",
" - Name background\n",
" - Click \"Create\" under \"Proteome file\" to choose where to save the background\n",
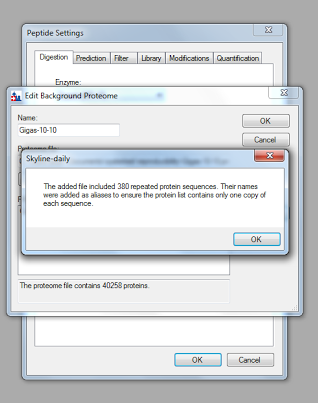
" - Click \"Add file\" under \"FASTA files\", and select your background proteome fasta file\n",
" - After the FASTA loads, Skyline may prompt you about deleting repeated protein sequences. Select \"OK\"\n",
" \n",
"  \n",
"\n",
"4. Select \"Trypsin [KR | P]\" under \"Enzyme\n",
"\n",
"
"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 2b: Add Background Proteome\n",
"\n",
"1. Settings >> Peptide Settings >> Digestion\n",
"2. Select \"Gigas-4-27\" under \"Background proteome\"\n",
"3. If \"Gigas-4-27\" is not an option\n",
" - Select \"Add\" under \"Background proteome\"\n",
" - Name background\n",
" - Click \"Create\" under \"Proteome file\" to choose where to save the background\n",
" - Click \"Add file\" under \"FASTA files\", and select your background proteome fasta file\n",
" - After the FASTA loads, Skyline may prompt you about deleting repeated protein sequences. Select \"OK\"\n",
" \n",
"  \n",
"\n",
"4. Select \"Trypsin [KR | P]\" under \"Enzyme\n",
"\n",
" "
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 2c: Adjust Peptide Settings\n",
"\n",
"1. Under the Prediction tab, make sure \"none\" is selected for retention time predictor\n",
"\n",
"
"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 2c: Adjust Peptide Settings\n",
"\n",
"1. Under the Prediction tab, make sure \"none\" is selected for retention time predictor\n",
"\n",
"  \n",
"\n",
"2. Under the Filter tab, click \"Auto-select all matching peptides\" and ensure 2 and 25 are the Min and Max lengths. Under \"Exclude N-Terminal AAs\" enter zero.\n",
"\n",
"
\n",
"\n",
"2. Under the Filter tab, click \"Auto-select all matching peptides\" and ensure 2 and 25 are the Min and Max lengths. Under \"Exclude N-Terminal AAs\" enter zero.\n",
"\n",
"  \n",
"\n",
"3. Under the Modification tab, select \"Carbamidomethyl (C)\" under structural modifications, \"heavy\" Isotope label type, and \"light\" Internal standard type.\n",
"\n",
"
\n",
"\n",
"3. Under the Modification tab, select \"Carbamidomethyl (C)\" under structural modifications, \"heavy\" Isotope label type, and \"light\" Internal standard type.\n",
"\n",
"  \n",
"\n",
"4. Use the following settings under the Quantification tab\n",
"\n",
"
\n",
"\n",
"4. Use the following settings under the Quantification tab\n",
"\n",
"  "
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 2d: Populate Analyte Tree\n",
"\n",
"1. Under File >> Import, select \"Transition List\"\n",
"2. Select the transition list (.csv) outlined in Step 1. This will populate the analyte tree only with targeted proteins and quality control (PRTC) peptides.\n",
"\n",
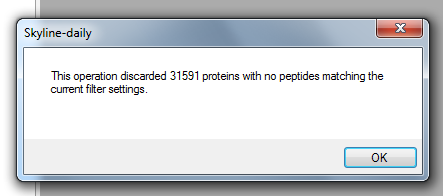
"Skyline will keep the proteins, peptides and transitions that match what it finds in the library provided in Step 2a. An alert, such as the one below, will show up to inform you how many proteins did not match the library. This is no cause for concern.\n",
"\n",
""
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 2e: Adjust Transition Settings\n",
"\n",
"1. **Settings >> Transition Settings >> Prediction**\n",
" - Precursor mass: Monoisotopic\n",
" - Product ion mass: Monoisotopic\n",
" - Collision energy: Thermo TSQ Vantage\n",
" - Declustering potential: None\n",
" - Optimization library: None\n",
" - Compensation voltage: None\n",
" - DO NOT select \"Use optimization values when present\"\n",
" \n",
"
"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 2d: Populate Analyte Tree\n",
"\n",
"1. Under File >> Import, select \"Transition List\"\n",
"2. Select the transition list (.csv) outlined in Step 1. This will populate the analyte tree only with targeted proteins and quality control (PRTC) peptides.\n",
"\n",
"Skyline will keep the proteins, peptides and transitions that match what it finds in the library provided in Step 2a. An alert, such as the one below, will show up to inform you how many proteins did not match the library. This is no cause for concern.\n",
"\n",
""
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 2e: Adjust Transition Settings\n",
"\n",
"1. **Settings >> Transition Settings >> Prediction**\n",
" - Precursor mass: Monoisotopic\n",
" - Product ion mass: Monoisotopic\n",
" - Collision energy: Thermo TSQ Vantage\n",
" - Declustering potential: None\n",
" - Optimization library: None\n",
" - Compensation voltage: None\n",
" - DO NOT select \"Use optimization values when present\"\n",
" \n",
"  \n",
"\n",
"2. **Settings >> Transition Settings >> Filter >> Peptides**\n",
" - Precursor charges: 2, 3\n",
" - Ion charges: 1, 2\n",
" - Ion types: y\n",
" - Product ion selection\n",
" - From: ion 2\n",
" - To: last ion\n",
" - DO NOT select any Special ions\n",
" - DO NOT specify any Precursor m/z exclusion window\n",
" - DO select \"Auto-select all matching transitions\"\n",
" \n",
"
\n",
"\n",
"2. **Settings >> Transition Settings >> Filter >> Peptides**\n",
" - Precursor charges: 2, 3\n",
" - Ion charges: 1, 2\n",
" - Ion types: y\n",
" - Product ion selection\n",
" - From: ion 2\n",
" - To: last ion\n",
" - DO NOT select any Special ions\n",
" - DO NOT specify any Precursor m/z exclusion window\n",
" - DO select \"Auto-select all matching transitions\"\n",
" \n",
"  \n",
" \n",
"3. **Transition Settings >> Library**\n",
" - Ion match tolerance: 0.5 m/z\n",
" - DO NOT select \"If a library spectrum is available, pick its most intense ions\"\n",
" \n",
"
\n",
" \n",
"3. **Transition Settings >> Library**\n",
" - Ion match tolerance: 0.5 m/z\n",
" - DO NOT select \"If a library spectrum is available, pick its most intense ions\"\n",
" \n",
"  \n",
" \n",
"4. **Transition Settings >> Instrument**\n",
" - Min m/z: 100\n",
" - Max m/z: 2000\n",
" - DO NOT select \"Dynamic min product m/z\"\n",
" - Method match tolerance m/z: 0.055 m/z\n",
" - DO NOT specify any other settings on this tab\n",
" \n",
"
\n",
" \n",
"4. **Transition Settings >> Instrument**\n",
" - Min m/z: 100\n",
" - Max m/z: 2000\n",
" - DO NOT select \"Dynamic min product m/z\"\n",
" - Method match tolerance m/z: 0.055 m/z\n",
" - DO NOT specify any other settings on this tab\n",
" \n",
"  \n",
" \n",
"5. **Transition Settings >> Full-Scan**\n",
" - Isotope peaks included: Count\n",
" - Precursor mass analyzer: Orbitrap\n",
" - Peaks: 3\n",
" - Resolving power: 60,000\n",
" - At: 400 m/z\n",
" - Isotope labeling enrichment: Default\n",
" - Acquisition method: Targeted\n",
" - Product mass analyzer: Centroided\n",
" - Isolation scheme: None\n",
" - Mass Accuracy: 20 ppm\n",
" - DO NOT select \"Use high-sensitivity extraction\"\n",
" - Select \"Use only scans within 2 minutes of MS/MS IDs\"\n",
" \n",
"
\n",
" \n",
"5. **Transition Settings >> Full-Scan**\n",
" - Isotope peaks included: Count\n",
" - Precursor mass analyzer: Orbitrap\n",
" - Peaks: 3\n",
" - Resolving power: 60,000\n",
" - At: 400 m/z\n",
" - Isotope labeling enrichment: Default\n",
" - Acquisition method: Targeted\n",
" - Product mass analyzer: Centroided\n",
" - Isolation scheme: None\n",
" - Mass Accuracy: 20 ppm\n",
" - DO NOT select \"Use high-sensitivity extraction\"\n",
" - Select \"Use only scans within 2 minutes of MS/MS IDs\"\n",
" \n",
"  "
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Step 3: Evaluate Dilution Curve\n",
"\n",
"The purpose of the dilution curve is to ensure the mass spectrometer methodology is specific to target proteins. Peptides that do not decrease in abundance when the concentration of protein loaded onto the machine decreases should be removed from analyses."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 3a: Import Data\n",
"\n",
"Under File > Import > Results, select \"Add single-injection replicates in files\". Navigate to the directory with RAW files named \"Dilution-Curve\". \n",
"\n",
"The dilution curve files are labelled \"D#-O,\" where \"D\" stands for \"dilution\", # represents [which dilution ratio was used](https://yaaminiv.github.io/SRM-Dilution-Calculations2/), and \"O\" stands for \"oyster.\" There are eight dilution files, so ensure all files are present before proceeding. \n",
"\n",
"**Select the files in the \"Dilution-Curve\" folder and click \"Open.\"**"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 3b: Clean Dilution Curve Data\n",
"\n",
"For each sample, go through the list of peptides and ensure Skyline selected the proper peak. You will need to click on the peptide name (the thing with the green dots next to them) in order to see the transition data. Use the [predicted retetion times](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-08-Final-Transitions/2017-07-11-Predicted-SRM-Retention-Times.xlsx) document to identify the correct peptide peak. Compare the average retention time predicted with the retention times in the Skyline document.\n",
"\n",
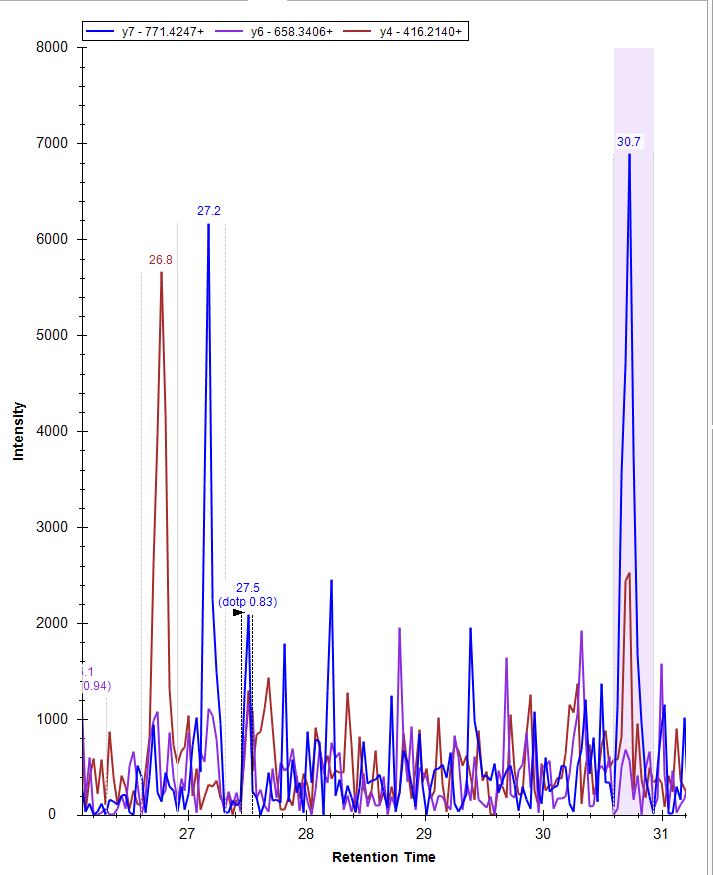
"The peak selected in Skyline will have a black arrow next to it. In the example below, Skyline has selected the peak with a retention time of 27.5. It also recognizes peaks with retention times of 26.8, 27.2 and 30.7.\n",
"\n",
"\n",
"\n",
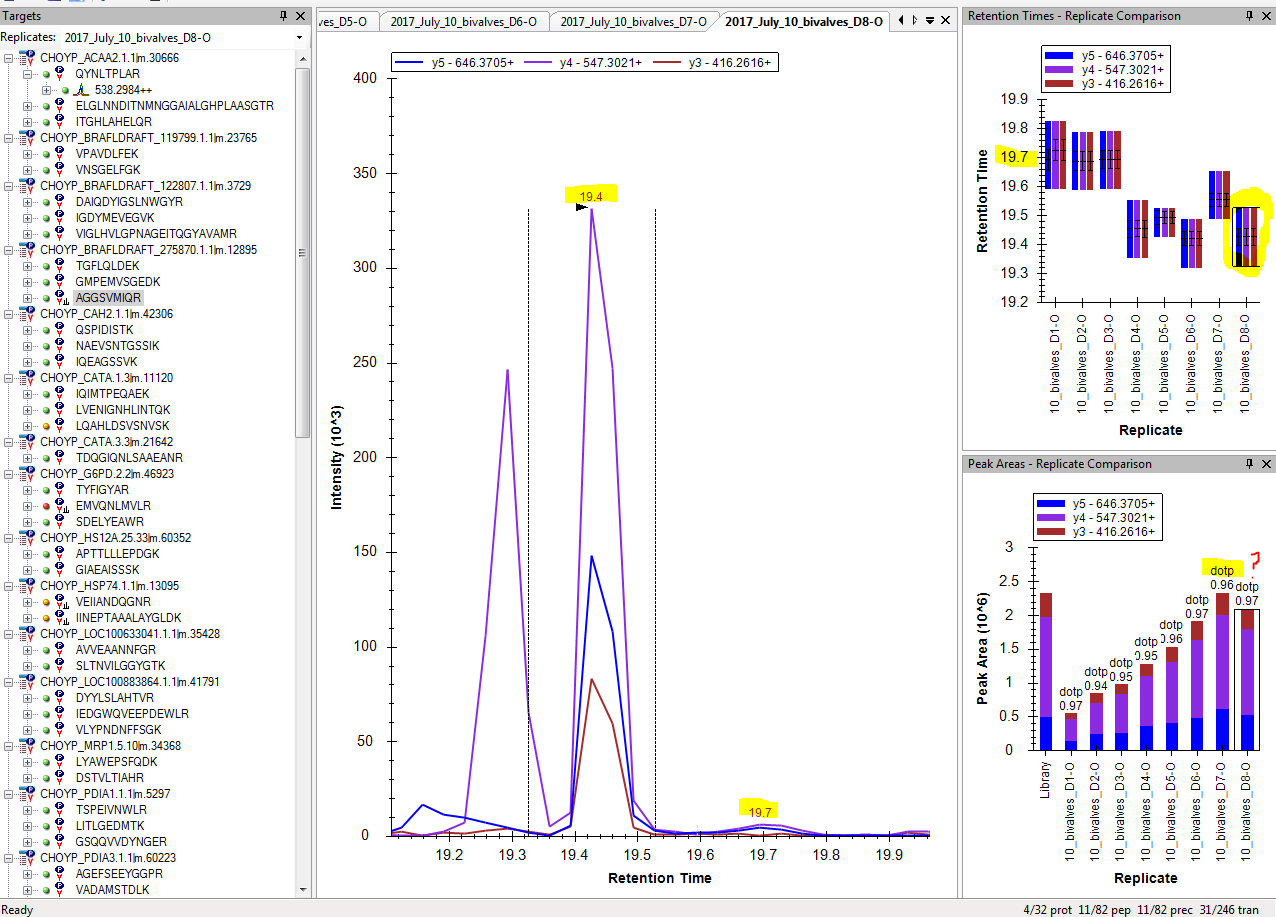
"In the screenshot below, there are two potential peaks that match the predicted retention time: 19.4 and 19.7\n",
"\n",
"\n",
"\n",
"In this instance, the peak at 19.4 is the correct peak. The correct peak will have all three transition fragments. Additionally, the correct retention time will be slightly earlier than the predicted retention time, since these dilution samples were run at the very end of our mass spectrometry run, and retention times get earlier as you keep running samples.\n",
"\n",
"Some more notes on Skyline features:\n",
"\n",
"- \"dotp\" is not something that's affecting the dilution curve\n",
"- The library bar in the Peak Area comparison does not affect the dilution curve either. It just means that Skyline recognizes this peptide and it matching it to information it finds in its own database. Some peptides have them, some don't. It's not important!\n",
"\n",
"If the correct peak is selected, adjust the peak boundaries (dashed lines) so they only surround the peak. This can be done simply by dragging the boundary to the desired ocation.\n",
"\n",
"If the incorrect peak is selected, you will need to manually select the correct peak. If Skyline recognizes the peak, simply click on the correct peak. If Skyline does not recognize the peak, hover over the x-axis (Retentiom time). The mouse will turn into a crosshair. Select the proper peak by dragging the crosshair over the retention times that the peak spans.\n",
"\n",
"If there is no discernable peak present (the transitions do not elute at the same time), right click and select \"Remove peak.\"\n",
"\n",
"After selecting the correct peak, right click and select \"Apply Peak to All.\" This may not correctly identify the proper peak for all samples, but for a good majority. You will still need to check all peptide peaks and adjust peak boundaries."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 3c: Identify excludable peptides\n",
"\n",
"Once the proper peaks are selected, examine peak areas across the dilution curve. D1-O has the highest concentration of oyster protein, and D8-O has the lowest concentration. As the concentration of protein loaded decreases, so should peak area:\n",
"\n",
"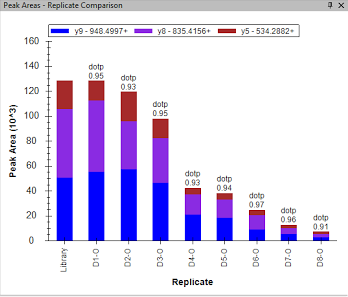\n",
"\n",
"If this is not the case, then the peptide needs to be exluced from analyses. Instructions on peptide exclusion will come later, so just make note of which peptides need to be removed for now.\n",
"\n",
"*The final dilution curve document can be found [here](http://owl.fish.washington.edu/spartina/DNR_SRM_20170728/Analyses/2017-09-06-GigasDilutionCurve.sky.zip). Transitions kept after dilution curve analyses should match those found in [this transition list](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-10-11-Final-SRM-Transition-List-with-PRTC.csv). Both of these files can be used for comparison.*"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Step 4: Create SRM Skyline Document\n",
"\n",
"This is the Skyline document used to view mass spectrometry data from oyster samples. This is also the primary document used for viewing sample data and assessing peak quality."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 4a: Duplicate the Dilution Curve Document\n",
"\n",
"Create a copy of the dilution curve Skyline document and rename it for SRM data."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 4b: Delete Peptides Based on Dilution Curve\n",
"\n",
"If there were any peptides that did not follow the expected pattern in the dilution curve, remove them from the analyte tree. To do this, right click on the peptide, then select \"Delete.\"\n",
"\n",
"Ensure all target proteins have at least two associated peptides before proceeding. If not, delete the protein from analysis as it no longer meets the criteria of a target."
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"### Step 4c: Add SRM Raw Data\n",
"\n",
"Under Edit >> Manage Results, remove all RAW files associated with the dilution curve. Under File > Import > Results, select \"Add single-injection replicates in files\". Navigate to the directory with RAW files, \"Oyster-Samples\". The files associated with samples are named \"2017_July_10_bivalves_NUMBER.\" \n",
"\n",
"Import all files in the \"Oyster-Samples\" folder, except for the one labelled \"2017_July_10_bivalves_QC18.\" Select files to import and click \"Open\"."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Step 5: Clean Data in Skyline\n",
"\n",
"In this step, files without any data will be removed and peaks in Skyline will be verfied against predicted retention times."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 5a: Remove Blanks\n",
"\n",
"Blank solutions without any oyster protein were run between sets of samples and should be excluded from analysis. Under Edit >> Manage Results, delete these files from the Skyline document. Blanks can be found using the [sample key](http://owl.fish.washington.edu/spartina/DNR_SRM_20170728/Analyses/2017-07-28-SRM-Sequence-File.xlsx). In the \"Comment\" column, \"blank\" indicates a file contains no oyster protein."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 5b: Remove Samples Without Any Data\n",
"\n",
"There could be sample files that did not pick up any peptide data whatsoever, even though they were not blanks. Thumb through the different sample results. If any samples display the phrase \"No product ion chromatograms found,\" for all peptides, remove the sample file from the document. \n",
"\n",
"
"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Step 3: Evaluate Dilution Curve\n",
"\n",
"The purpose of the dilution curve is to ensure the mass spectrometer methodology is specific to target proteins. Peptides that do not decrease in abundance when the concentration of protein loaded onto the machine decreases should be removed from analyses."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 3a: Import Data\n",
"\n",
"Under File > Import > Results, select \"Add single-injection replicates in files\". Navigate to the directory with RAW files named \"Dilution-Curve\". \n",
"\n",
"The dilution curve files are labelled \"D#-O,\" where \"D\" stands for \"dilution\", # represents [which dilution ratio was used](https://yaaminiv.github.io/SRM-Dilution-Calculations2/), and \"O\" stands for \"oyster.\" There are eight dilution files, so ensure all files are present before proceeding. \n",
"\n",
"**Select the files in the \"Dilution-Curve\" folder and click \"Open.\"**"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 3b: Clean Dilution Curve Data\n",
"\n",
"For each sample, go through the list of peptides and ensure Skyline selected the proper peak. You will need to click on the peptide name (the thing with the green dots next to them) in order to see the transition data. Use the [predicted retetion times](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-08-Final-Transitions/2017-07-11-Predicted-SRM-Retention-Times.xlsx) document to identify the correct peptide peak. Compare the average retention time predicted with the retention times in the Skyline document.\n",
"\n",
"The peak selected in Skyline will have a black arrow next to it. In the example below, Skyline has selected the peak with a retention time of 27.5. It also recognizes peaks with retention times of 26.8, 27.2 and 30.7.\n",
"\n",
"\n",
"\n",
"In the screenshot below, there are two potential peaks that match the predicted retention time: 19.4 and 19.7\n",
"\n",
"\n",
"\n",
"In this instance, the peak at 19.4 is the correct peak. The correct peak will have all three transition fragments. Additionally, the correct retention time will be slightly earlier than the predicted retention time, since these dilution samples were run at the very end of our mass spectrometry run, and retention times get earlier as you keep running samples.\n",
"\n",
"Some more notes on Skyline features:\n",
"\n",
"- \"dotp\" is not something that's affecting the dilution curve\n",
"- The library bar in the Peak Area comparison does not affect the dilution curve either. It just means that Skyline recognizes this peptide and it matching it to information it finds in its own database. Some peptides have them, some don't. It's not important!\n",
"\n",
"If the correct peak is selected, adjust the peak boundaries (dashed lines) so they only surround the peak. This can be done simply by dragging the boundary to the desired ocation.\n",
"\n",
"If the incorrect peak is selected, you will need to manually select the correct peak. If Skyline recognizes the peak, simply click on the correct peak. If Skyline does not recognize the peak, hover over the x-axis (Retentiom time). The mouse will turn into a crosshair. Select the proper peak by dragging the crosshair over the retention times that the peak spans.\n",
"\n",
"If there is no discernable peak present (the transitions do not elute at the same time), right click and select \"Remove peak.\"\n",
"\n",
"After selecting the correct peak, right click and select \"Apply Peak to All.\" This may not correctly identify the proper peak for all samples, but for a good majority. You will still need to check all peptide peaks and adjust peak boundaries."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 3c: Identify excludable peptides\n",
"\n",
"Once the proper peaks are selected, examine peak areas across the dilution curve. D1-O has the highest concentration of oyster protein, and D8-O has the lowest concentration. As the concentration of protein loaded decreases, so should peak area:\n",
"\n",
"\n",
"\n",
"If this is not the case, then the peptide needs to be exluced from analyses. Instructions on peptide exclusion will come later, so just make note of which peptides need to be removed for now.\n",
"\n",
"*The final dilution curve document can be found [here](http://owl.fish.washington.edu/spartina/DNR_SRM_20170728/Analyses/2017-09-06-GigasDilutionCurve.sky.zip). Transitions kept after dilution curve analyses should match those found in [this transition list](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-10-11-Final-SRM-Transition-List-with-PRTC.csv). Both of these files can be used for comparison.*"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Step 4: Create SRM Skyline Document\n",
"\n",
"This is the Skyline document used to view mass spectrometry data from oyster samples. This is also the primary document used for viewing sample data and assessing peak quality."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 4a: Duplicate the Dilution Curve Document\n",
"\n",
"Create a copy of the dilution curve Skyline document and rename it for SRM data."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 4b: Delete Peptides Based on Dilution Curve\n",
"\n",
"If there were any peptides that did not follow the expected pattern in the dilution curve, remove them from the analyte tree. To do this, right click on the peptide, then select \"Delete.\"\n",
"\n",
"Ensure all target proteins have at least two associated peptides before proceeding. If not, delete the protein from analysis as it no longer meets the criteria of a target."
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"### Step 4c: Add SRM Raw Data\n",
"\n",
"Under Edit >> Manage Results, remove all RAW files associated with the dilution curve. Under File > Import > Results, select \"Add single-injection replicates in files\". Navigate to the directory with RAW files, \"Oyster-Samples\". The files associated with samples are named \"2017_July_10_bivalves_NUMBER.\" \n",
"\n",
"Import all files in the \"Oyster-Samples\" folder, except for the one labelled \"2017_July_10_bivalves_QC18.\" Select files to import and click \"Open\"."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Step 5: Clean Data in Skyline\n",
"\n",
"In this step, files without any data will be removed and peaks in Skyline will be verfied against predicted retention times."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 5a: Remove Blanks\n",
"\n",
"Blank solutions without any oyster protein were run between sets of samples and should be excluded from analysis. Under Edit >> Manage Results, delete these files from the Skyline document. Blanks can be found using the [sample key](http://owl.fish.washington.edu/spartina/DNR_SRM_20170728/Analyses/2017-07-28-SRM-Sequence-File.xlsx). In the \"Comment\" column, \"blank\" indicates a file contains no oyster protein."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 5b: Remove Samples Without Any Data\n",
"\n",
"There could be sample files that did not pick up any peptide data whatsoever, even though they were not blanks. Thumb through the different sample results. If any samples display the phrase \"No product ion chromatograms found,\" for all peptides, remove the sample file from the document. \n",
"\n",
" \n",
"\n",
"Using the [sample key](http://owl.fish.washington.edu/spartina/DNR_SRM_20170728/Analyses/2017-07-28-SRM-Sequence-File.xlsx), identify that file's technical replicate and delete is as well. Technical replicates will have the same oyster ID in the \"Comment\" column (ex. O01-1 and O01-2)."
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"### Step 5c: Select Proper Peaks and Fix Boundaries\n",
"\n",
"Similar to the dilution curve, all peptide peaks need to be verified for each sample. See Step 3a for instructions.\n",
"\n",
"*The final Skyline document can be found [here](http://owl.fish.washington.edu/spartina/DNR_SRM_20170728/Analyses/2017-09-12-Gigas-SRM-ReplicatesOnly-PostDilutionCurve-RevisedSettings.sky.zip) for comparison.*"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Step 6: Export Data\n",
"\n",
"To proceed with downstream analyses, data must be exported from Skyline as a .csv."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Under File > Export > Report, use the following settings to export Skyline data as a .csv.\n",
"\n",
"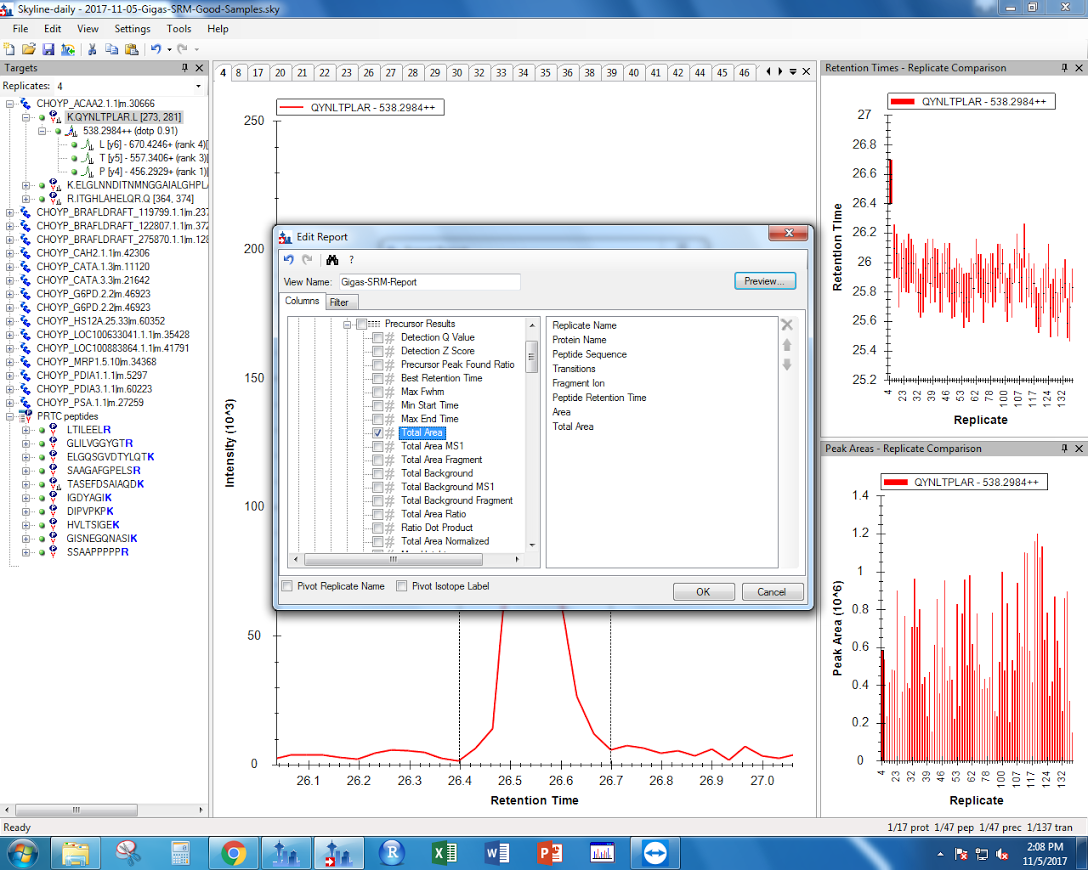\n",
"\n",
"If you click preview, this is what the document should look like:\n",
"\n",
"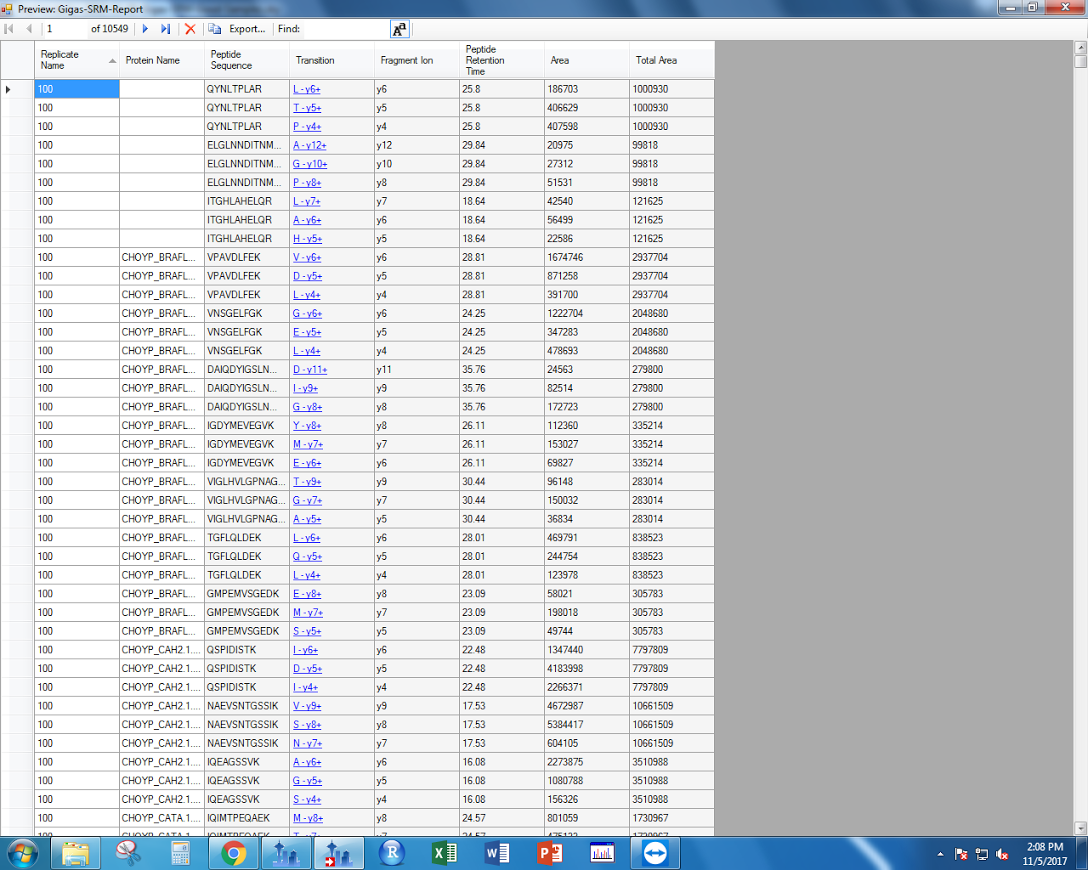\n",
"\n",
"*Exported data can be found [here](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-10-10-Troubleshooting/2017-11-05-Integrated-Dataset/2017-11-05-Gigas-SRM-Good-Samples-Total-Area.csv) for comparison*."
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"## Step 7: Analyze Technical Replication\n",
"\n",
"Each oyster sample was run in technical duplicates. Oyster protein sasmples [prepared](https://github.com/RobertsLab/resources/blob/master/protocols/ProteinprepforMSMS.md) prior to mass spectrometry were placed in individual autosampler vials. Each vial was injected twice, producing two technical replicates. Before proceeding, it is important to verify that technical replicates have the same peak area for all peptides and transitions. If this is not the case, those samples must be removed from the analysis."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 7a: Collect Materials\n",
"\n",
"- [Skyline data](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-09-12-Gigas-SRM-ReplicatesOnly-PostDilutionCurve-NoPivot-RevisedSettings-Report.csv)\n",
"- [Sample key (.xlsx)](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-07-28-SRM-Sequence-File.xlsx)\n",
"- [Biological Replication Key](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-09-06-Biological-Replicate-Information.csv)"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 7b: Create a Sequence File (.csv)\n",
"\n",
"Duplicate the [sample key (.xlsx)](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-07-28-SRM-Sequence-File.xlsx), and remove all rows that are blanks, QCs or for the dilution curve. Also remove rows for samples that had no data Step 5b and their technical replicates. Save this file as a .csv.\n",
"\n",
"*The sequence file I generated can be found [here](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-07-28-SRM-Samples-Sequence-File.csv) for comparison*."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 7b: Analyze in R Studio\n",
"\n",
"Using this [R script](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-10-10-Troubleshooting/2017-11-05-Integrated-Dataset/2017-11-05-NMDS-for-Technical-Replication.R), analyze technical replication."
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"## Step 8: Analyze Biological Replication\n",
"\n",
"Once technical replication is verified, NMDS and ANOSIM analyses can be used to examine similarities in protein abundance between site and eelgrass condition."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 8a: Collect Materials\n",
"\n",
"- Same materials needed as previous step."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 8b: Analyze in R Studio\n",
"\n",
"Using this [R script](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-10-10-Troubleshooting/2017-11-05-Integrated-Dataset/2017-11-05-NMDS-ANOSIM-for-Cluster-Analysis.R), run ANOSIM on averaged technical replicate data."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 8c: Analyze Protein Functions\n",
"\n",
"Using this [R script](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-10-10-Troubleshooting/2017-11-05-Integrated-Dataset/2017-11-05-NMDS-ANOSIM-for-Individual-Proteins.R), run ANOSIM analyses on proteins groups together by similar functions"
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"## Step 9: Visualize Data\n",
"\n",
"This step lays out different methods for visualizing protein abundance data."
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"### Step 9a: Collect Materials\n",
"\n",
"- Completion of previous [R script for technical replication](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-09-06-NMDS-for-Technical-Replication.R). All products and datasets should still be in the workspace."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 9b: Analyze in R Studio"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Using this [R script](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-10-10-Troubleshooting/2017-11-05-Integrated-Dataset/2017-11-06-Boxplots/2017-11-06-Protein-Area-Boxplots-after-Integration.R), create boxplots for protein areas across sites. The script also runs one-way ANOVA, post-hoc Tukey's Honest Significant Difference tests, and power analyses on dataset."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"*Next steps: ROC curves, plotting peak area on the y, amount of peptide (moles) on the x.*"
]
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
}
],
"metadata": {
"anaconda-cloud": {},
"kernelspec": {
"display_name": "Python [conda root]",
"language": "python",
"name": "conda-root-py"
},
"language_info": {
"codemirror_mode": {
"name": "ipython",
"version": 3
},
"file_extension": ".py",
"mimetype": "text/x-python",
"name": "python",
"nbconvert_exporter": "python",
"pygments_lexer": "ipython3",
"version": "3.5.2"
}
},
"nbformat": 4,
"nbformat_minor": 1
}
\n",
"\n",
"Using the [sample key](http://owl.fish.washington.edu/spartina/DNR_SRM_20170728/Analyses/2017-07-28-SRM-Sequence-File.xlsx), identify that file's technical replicate and delete is as well. Technical replicates will have the same oyster ID in the \"Comment\" column (ex. O01-1 and O01-2)."
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"### Step 5c: Select Proper Peaks and Fix Boundaries\n",
"\n",
"Similar to the dilution curve, all peptide peaks need to be verified for each sample. See Step 3a for instructions.\n",
"\n",
"*The final Skyline document can be found [here](http://owl.fish.washington.edu/spartina/DNR_SRM_20170728/Analyses/2017-09-12-Gigas-SRM-ReplicatesOnly-PostDilutionCurve-RevisedSettings.sky.zip) for comparison.*"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Step 6: Export Data\n",
"\n",
"To proceed with downstream analyses, data must be exported from Skyline as a .csv."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Under File > Export > Report, use the following settings to export Skyline data as a .csv.\n",
"\n",
"\n",
"\n",
"If you click preview, this is what the document should look like:\n",
"\n",
"\n",
"\n",
"*Exported data can be found [here](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-10-10-Troubleshooting/2017-11-05-Integrated-Dataset/2017-11-05-Gigas-SRM-Good-Samples-Total-Area.csv) for comparison*."
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"## Step 7: Analyze Technical Replication\n",
"\n",
"Each oyster sample was run in technical duplicates. Oyster protein sasmples [prepared](https://github.com/RobertsLab/resources/blob/master/protocols/ProteinprepforMSMS.md) prior to mass spectrometry were placed in individual autosampler vials. Each vial was injected twice, producing two technical replicates. Before proceeding, it is important to verify that technical replicates have the same peak area for all peptides and transitions. If this is not the case, those samples must be removed from the analysis."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 7a: Collect Materials\n",
"\n",
"- [Skyline data](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-09-12-Gigas-SRM-ReplicatesOnly-PostDilutionCurve-NoPivot-RevisedSettings-Report.csv)\n",
"- [Sample key (.xlsx)](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-07-28-SRM-Sequence-File.xlsx)\n",
"- [Biological Replication Key](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-09-06-Biological-Replicate-Information.csv)"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 7b: Create a Sequence File (.csv)\n",
"\n",
"Duplicate the [sample key (.xlsx)](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-07-28-SRM-Sequence-File.xlsx), and remove all rows that are blanks, QCs or for the dilution curve. Also remove rows for samples that had no data Step 5b and their technical replicates. Save this file as a .csv.\n",
"\n",
"*The sequence file I generated can be found [here](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-07-28-SRM-Samples-Sequence-File.csv) for comparison*."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 7b: Analyze in R Studio\n",
"\n",
"Using this [R script](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-10-10-Troubleshooting/2017-11-05-Integrated-Dataset/2017-11-05-NMDS-for-Technical-Replication.R), analyze technical replication."
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"## Step 8: Analyze Biological Replication\n",
"\n",
"Once technical replication is verified, NMDS and ANOSIM analyses can be used to examine similarities in protein abundance between site and eelgrass condition."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 8a: Collect Materials\n",
"\n",
"- Same materials needed as previous step."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 8b: Analyze in R Studio\n",
"\n",
"Using this [R script](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-10-10-Troubleshooting/2017-11-05-Integrated-Dataset/2017-11-05-NMDS-ANOSIM-for-Cluster-Analysis.R), run ANOSIM on averaged technical replicate data."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 8c: Analyze Protein Functions\n",
"\n",
"Using this [R script](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-10-10-Troubleshooting/2017-11-05-Integrated-Dataset/2017-11-05-NMDS-ANOSIM-for-Individual-Proteins.R), run ANOSIM analyses on proteins groups together by similar functions"
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"## Step 9: Visualize Data\n",
"\n",
"This step lays out different methods for visualizing protein abundance data."
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"### Step 9a: Collect Materials\n",
"\n",
"- Completion of previous [R script for technical replication](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-09-06-NMDS-for-Technical-Replication.R). All products and datasets should still be in the workspace."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Step 9b: Analyze in R Studio"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Using this [R script](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_SRM_20170902/2017-10-10-Troubleshooting/2017-11-05-Integrated-Dataset/2017-11-06-Boxplots/2017-11-06-Protein-Area-Boxplots-after-Integration.R), create boxplots for protein areas across sites. The script also runs one-way ANOVA, post-hoc Tukey's Honest Significant Difference tests, and power analyses on dataset."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"*Next steps: ROC curves, plotting peak area on the y, amount of peptide (moles) on the x.*"
]
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
}
],
"metadata": {
"anaconda-cloud": {},
"kernelspec": {
"display_name": "Python [conda root]",
"language": "python",
"name": "conda-root-py"
},
"language_info": {
"codemirror_mode": {
"name": "ipython",
"version": 3
},
"file_extension": ".py",
"mimetype": "text/x-python",
"name": "python",
"nbconvert_exporter": "python",
"pygments_lexer": "ipython3",
"version": "3.5.2"
}
},
"nbformat": 4,
"nbformat_minor": 1
}