{
"cells": [
{
"cell_type": "markdown",
"metadata": {},
"source": [
"# SRM Target Identification in Skyline"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Now that I have a [short list of proteins](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-07-Preliminary-Transitions/2017-07-07-Protein-Shortlist-Evalues.csv), I can examine peaks in Skyline and select targets for our SRM assay starting June 10, 2017. The goal is to have 100-150 transitions total. Assuming I can find three good transitions for every peptide, and that each protein has three good peptides, I need 12-16 proteins total."
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"## Step 1: Create a Duplicate Skyline Document"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"I don't want to overwrite my full Skyline document, so I will make a new one. To do this, I first opened up my [current Skyline document](http://owl.fish.washington.edu/spartina/DNR_Skyline_20170524/Gigas-6-10-DIA.sky.zip). I then went to File >> Save As and named my new file:\n",
"\n",
"\n",
"\n",
"I closed out my old file and opened the [new Skyline document](http://owl.fish.washington.edu/spartina/DNR_Skyline_SRM_20170707/Gigas-7-7-Transition-List.sky.zip):\n",
"\n",
""
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Step 2: Evaluate Peak Quality"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Peak Deletion\n",
"\n",
"In the new Skyline document, I went through proteins selected on my [short list](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-07-Preliminary-Transitions/2017-07-07-Protein-Shortlist-Evalues.csv) and looked at peak quality. I removed proteins, peptides and transitions on the shortlist based on the criteria in [Emma's Skyline tutorial slides](https://github.com/RobertsLab/project-pacific.oyster-larvae/blob/master/Skyline-example-files-ETS.sky/slides01.pdf).\n",
"\n",
"Here are the proteins I deemed first priority after talking to Steven:\n",
"\n",
"- CHOYP_BRAFLDRAFT_119799.1.1|m.23765\n",
"- CHOYP_BRAFLDRAFT_91636.1.11|m.28449\n",
"- CHOYP_CAT.1.1|m.57038\n",
"- CHOYP_CATA.2.3|m.18454\n",
"- CHOYP_DNJA2.2.2|m.51791\n",
"- CHOYP_G6PD.2.2|m.46923\n",
"- CHOYP_GPX4-A.1.1|m.257\n",
"- CHOYP_HSPA12A.10.27|m.35252\n",
"- CHOYP_HSPA12A.19.27|m.54978\n",
"- CHOYP_HYOU1.2.3|m.12568\n",
"- CHOYP_LOC100371830.4.6|m.25343\n",
"- CHOYP_LOC100377195.2.2|m.61720\n",
"- CHOYP_LOC100150876.1.1|m.61100\n",
"- CHOYP_LOC101169658.1.3|m.18820\n",
"- CHOYP_LOC576041.1.1|m.28220\n",
"- CHOYP_MANL.8.9|m.48353\n",
"- CHOYP_MRP1.5.10|m.34368\n",
"\n",
"**Deletion criteria**:\n",
"\n",
"- Delete a protein if it only has one associated peptide\n",
"- Delete a peptide if\n",
" - There are less than three transitions\n",
" - There is too much peak intereference, so the peak isn't clearly defined (i.e. a sloppy peak)\n",
" - There is missing data for samples\n",
"- Delete a transition if\n",
" - It is a precursor ion\n",
" - It has low abundance (want to keep the three most abundant transitions)\n",
" - It is noisy\n",
" \n",
"For example, the least abundant transition in this peak (highlighted in red) is noisy...\n",
"\n",
"\n",
"\n",
"...so I deleted it.\n",
"\n",
"\n",
"\n",
"The first photo shows what this peak looks like in oyster 1, while the second photo shows the same peak after transition deletion in oyster 19. Because peak quality varies between samples, it is important to check that the peak is consistently good between all of my replicates. While the peak looks good in oyster 1, there is some slight intereference at the right end of the peak in oyster 19."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Boundary Refinement\n",
"\n",
"Just like when [error checking](https://yaaminiv.github.io/Skyline-Error-Checking-Round2/), I adjusted peak boundaries if they weren't correct to ensure I had the right peak retention times."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Protein Selection\n",
"\n",
"The following proteins from my shortlist did not have good quality peaks in Skyline:\n",
"\n",
"- CHOYP_HYOU1.2.3|m.12568\n",
"- CHOYP_GPX4-A.1.1|m.257\n",
"- CHOYP_BRAFLDRAFT_91636.1.11|m.28449\n",
"- CHOYP_LOC576041.1.1|m.28220\n",
"- CHOYP_HSPA12A.19.27|m.54978\n",
"- CHOYP_HSPA12A.10.27|m.35252\n",
"- CHOYP_LOC100371830.4.6|m.25343\n",
"\n",
"Using their annotations, I looked for similar proteins in my short list. I found the following substitutes with decent quality peaks:\n",
"\n",
"- CHOYP_ACAA2.1.1|m.30666\n",
"- CHOYP_BRAFLDRAFT_122807.1.1|m.3729\n",
"- CHOYP_BRAFLDRAFT_275870.1.1|m.12895\n",
"- CHOYP_BRAFLDRAFT_88228.1.1|m.51515\n",
"- CHOYP_CAH1.1.2|m.24247\n",
"- CHOYP_CAH2.1.1|m.42306\n",
"- CHOYP_EF2.1.5|m.1541\n",
"- CHOYP_HS12A.11.33|m.39341\n",
"- CHOYP_HS12A.14.33|m.41988\n",
"- CHOYP_HS12A.21.33|m.58427\n",
"- CHOYP_HS12A.25.33|m.60352\n",
"- CHOYP_LOC100883864.1.1|m.41791\n",
"- CHOYP_LOC594665.1.1|m.8448\n",
"- CHOYP_PDIA1.1.1|m.5297"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Step 3: Export Preliminary Transition List"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"After sorting through the short list, I had 23 proteins, with 64 peptides and 228 transitions. My goal was to have proteins with three peptides each, and each of those peptides would have three associated transitions. However, there were some proteins with only two good peptides, and some peptides had more than three good transitions. One protein, CHOYP_LOC594665.1.1|m.8448, only had one good peptide. I only kept it because it was Extracellular superoxide dismutase. While it is unlikely that Skyline can recognize a protein by just one peptide, I figured I would ask Emma."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"To export the preliminary transition list, I went to File >> Export >> Transition List.\n",
"\n",
"\n",
"\n",
"I changed the \"Instrument Type\" to Thermo and exported the list.\n",
"\n",
"\n",
"\n",
"After I had my [preliminary transition list](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-07-Preliminary-Transitions/2017-07-07-Preliminary-Target-Transitions.csv), I used [the R script](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-07-Preliminary-Transitions/2017-07-05-Differentially-Expressed-Annotations.R) to merge transitions with e-values. I sent the [transitions with e-values](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-07-Preliminary-Transitions/2017-07-07-Preliminary-Target-Transitions-Evalues.csv) to Steven and Emma for feedback to help narrow down transitions even further."
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"## Update: 2017-07-08"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Emma's Feedback\n",
"\n",
"Just as I thought, having one peptide for a protein will not be enough to confidently say that peptide is truly from that protein. Therefore, I will need to delete Extracellular superoxide dismutase from my protein list. After looking at my transitions, Emma noticed that some of my lower abundance transitions were not the greatest.\n",
"\n",
"For example, the transition highlighed in red below has a much lower intensity than the others:\n",
"\n",
"\n",
"\n",
"And the fifth transition in this peptide isn't even visible:\n",
"\n",
"\n",
"\n",
"However, Emma also said that SRM is good at detecting low abundance peptides. It is up to me to decide whether or not I want them. She also pointed out that I had a few peaks that had significance intereference and were pretty sloppy:\n",
"\n",
"\n",
"\n",
"Peaks like the one above were deleted immediately."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Steven asked that I refine my transitions such that I have an excess of six proteins. After this, he can go through and pick the most interesting ones."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Step 4: Refine Transition List"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"I documented my process in [this Excel document](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-08-Final-Transitions/2017-07-08-Target-Selection-Process-Notes.csv) because it was convenient. Here's what I did.\n",
"\n",
"To revise my transitions, I first deleted sloppy peptides and any protein that only had one peptide associated with it.\n",
"\n",
" \n"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"I then checked my original Skyline document for any higher quality peptides from my low-quality proteins. I only found one suitable replacement.\n",
"\n",
"
\n"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"I then checked my original Skyline document for any higher quality peptides from my low-quality proteins. I only found one suitable replacement.\n",
"\n",
" "
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"I also searched for backups in my shortlist and longlist.\n",
"\n",
"
"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"I also searched for backups in my shortlist and longlist.\n",
"\n",
" "
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Using the same criteria in Step 2, I evaluated my proteins.\n",
"\n",
"
"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Using the same criteria in Step 2, I evaluated my proteins.\n",
"\n",
" "
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"I sent the following proteins for a final review by Steven and Emma, leading to 134 transitions:\n",
" \n",
"
"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"I sent the following proteins for a final review by Steven and Emma, leading to 134 transitions:\n",
" \n",
" \n",
"\n",
"[Revised Skyline document](http://owl.fish.washington.edu/spartina/DNR_Skyline_SRM_20170707/Gigas-7-8-Revised-Transition-List.sky.zip)"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Update 2017-07-09"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Steven's Feedback\n",
"\n",
"Steven suggested that I hit 150 transitions and max out our capacity. However, I've already pored over both the short and long protein lists. He offered to help me search for proteins in the full Skyline output. I merged [my full Skyline output](http://owl.fish.washington.edu/spartina/DNR_Skyline_20170524/2017-06-10-protein-areas-only-error-checked.csv) with annotations to get [this list](https://raw.githubusercontent.com/RobertsLab/project-oyster-oa/master/analyses/DNR_Skyline_20170524/2017-07-09-Full-Skyline-Output-Annotations.csv). I evaluated the following proteins that Steven identified as interesting:\n",
"\n",
"
\n",
"\n",
"[Revised Skyline document](http://owl.fish.washington.edu/spartina/DNR_Skyline_SRM_20170707/Gigas-7-8-Revised-Transition-List.sky.zip)"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Update 2017-07-09"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Steven's Feedback\n",
"\n",
"Steven suggested that I hit 150 transitions and max out our capacity. However, I've already pored over both the short and long protein lists. He offered to help me search for proteins in the full Skyline output. I merged [my full Skyline output](http://owl.fish.washington.edu/spartina/DNR_Skyline_20170524/2017-06-10-protein-areas-only-error-checked.csv) with annotations to get [this list](https://raw.githubusercontent.com/RobertsLab/project-oyster-oa/master/analyses/DNR_Skyline_20170524/2017-07-09-Full-Skyline-Output-Annotations.csv). I evaluated the following proteins that Steven identified as interesting:\n",
"\n",
" \n",
"\n",
"
\n",
"\n",
" \n",
"\n",
"Most of the peaks had interference or missing data, but there were a handful I added to my protein list. Just to recap, here's what I was looking for.\n",
"\n",
"\n",
"\n",
"\n",
"\n",
"**Figures 1-2**. A noisy transition in Figure 1 (highlighted in red) that when deleted, produces a much better looking peak in Figure 2.\n",
"\n",
"\n",
"\n",
"**Figure 3**. A peak showing slight interference. I avoided this when possible. When interference was more extreme, I deleted the peptide.\n",
"\n",
"\n",
"\n",
"**Figure 4**. An example of an ideal peak."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"I then evaluated all of my proteins one last time:\n",
"\n",
"
\n",
"\n",
"Most of the peaks had interference or missing data, but there were a handful I added to my protein list. Just to recap, here's what I was looking for.\n",
"\n",
"\n",
"\n",
"\n",
"\n",
"**Figures 1-2**. A noisy transition in Figure 1 (highlighted in red) that when deleted, produces a much better looking peak in Figure 2.\n",
"\n",
"\n",
"\n",
"**Figure 3**. A peak showing slight interference. I avoided this when possible. When interference was more extreme, I deleted the peptide.\n",
"\n",
"\n",
"\n",
"**Figure 4**. An example of an ideal peak."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"I then evaluated all of my proteins one last time:\n",
"\n",
" \n",
"\n",
"And I finally had a list of 146 targets from 16 proteins!\n",
"\n",
"
\n",
"\n",
"And I finally had a list of 146 targets from 16 proteins!\n",
"\n",
" "
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Final Transition List\n",
"\n",
"[Final transitions](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-08-Final-Transitions/2017-07-09-Final-SRM-Transitions.csv)\n",
"\n",
"[Final Skyline document](http://owl.fish.washington.edu/spartina/DNR_Skyline_SRM_20170707/Gigas-7-9-Final-SRM-Transitions.sky.zip)"
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"## Update 2017-07-10"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Emma forgot that the 150 transition target maximum needs to include PRTC peptides! There are 38 transitions from PRTC peptides that we added, so we needed to cut down on my transition targets.\n",
"\n",
"The first thing we did was look at the final target list:\n",
"\n",
"\n",
"\n",
"In this list, there are proteins that have more than three target transitions per peptide. For the peptides with more than three associated transitions, we removed the least abundant ones so that we had a total of three transitions at the end. Additionally, the two catalase proteins, CHOYPCATA1.3|m.1120 and CHOYPCATA3.3|m.21642, had two peptides associated with both of them, LVENIGNHLINTQK and LQAHLDSVSNVSK. We deleted the redundant listing of these peptides.\n",
"\n",
"After this, we still needed to reduce the number of transitions, so I referred to the notes I had from my final review:\n",
"\n",
"\n",
"\n",
"There were two proteins that had one slightly sloppy peptide each, so we deleted them. Our [revised final transition list](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-08-Final-Transitions/2017-07-10-SRM-Transitions-With-PRTC.csv) has 159 total transitions. The 150 transition maximum is there because the machine can reliably detect 150 transitions at a time. We're pushing it with 159, so we will check our data to ensure our method is good. I also [revised my Skyline document](http://owl.fish.washington.edu/spartina/DNR_Skyline_SRM_20170707/2017-07-10-FINAL-SRM-Transitions-with-PRTC/Gigas-7-10-Final-Transition-List.sky.zip) to reflect these changes."
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"## Update 2017-07-11"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Predicted Retention Time Calculations\n",
"\n",
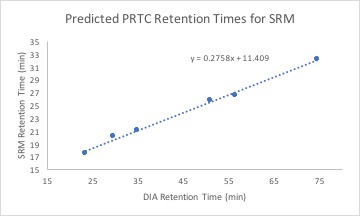
"Now that the [SRM assay is running](https://yaaminiv.github.io/SRM-Assay-Day1/), I need to confirm that I am getting the data I want. To do this, Emma and I selected a few PRTC peptides to find retention times for:\n",
"\n",
"- LTILEELR\n",
"- ELGQSGVDTYLQTK\n",
"- SAAGAFGPELSR\n",
"- DIPVPKPK\n",
"- HVLTSIGEK\n",
"- SSAAPPPPPR\n",
"\n",
"Using the fifth SRM QC run, I got retention times for these peptides. Emma went through 2 oyster DIA files (oyster 5 and oyster 10) and one geoduck DIA file's data and found retention times for our previous mass spectrometer run. I then regressed the SRM retention times against the DIA retention times to get a linear equation.\n",
"\n",
"\n",
"\n",
"Using this equation and DIA retention times for my transition targets, I calculated predicted SRM retention times. My data can be found [here](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-08-Final-Transitions/2017-07-11-Predicted-SRM-Retention-Times.xlsx). "
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Retention Time Confirmation\n",
"\n",
"In Skyline, I went through each peptide in two of my newly-collected oyster samples to ensure that Skyline picked the right peak. While the peaks were present around their calculation retention time, Skyline did not pick that peak for all but a few peptides.\n",
"\n",
"For example, Skyline chose a peak at 34.4 min for this peptide, when the best peak is clearly around 28 minutes. The best peak matches the best retention time I calculated as well.\n",
"\n",
"\n",
"\n",
"To fix this, I clicked on the peak I wanted Skyline to measure. Once I clicked on it, a black arrow appeared next to it.\n",
"\n",
"\n",
"\n",
"I then right-clicked and selected \"Apply Peak to All\", which had Skyline select the best peak for all samples in the document. Below, you can see that both samples have peaks made of three coeluting fragments around 28 minutes.\n",
"\n",
"\n",
"\n",
"I went through this process for all of my peaks. While Skyline may not be fully capable of picking the best peaks, the peaks were present! This is confirmation that the SRM assay works."
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"## Update 2017-07-18"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Since I have some time before I need to analyze my SRM data, I wanted to find ways to visualize my final transition list. The past few times I made NMDS plots, heatmaps and REVIGO plots, so I figured I would do the same this time. I used code [in this R script](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-08-Final-Transitions/2017-07-18-Final-Transition-Visualizations.R)."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
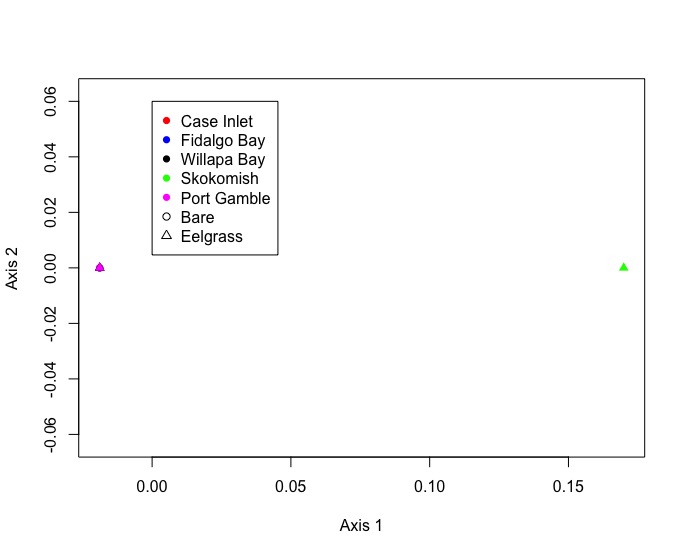
"### NMDS plot\n",
"\n",
"\n",
"\n",
"I ended up with all but one site and eelgrass condition nested on top of eachother. Not entire sure waht this means. I'll need to consult Emma to ensure that I did this correctly."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
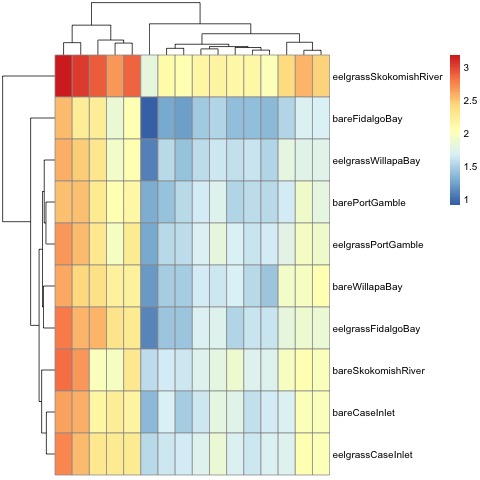
"### Heatmap\n",
"\n",
"\n",
"\n",
"My heatmap is a much cleaner way to visualize the proteins I selected for my final transition list, and how the expression differs between sites."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### REVIGO\n",
"\n",
"After I exported a list of [proteins, GOterms and p-values](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-08-Final-Transitions/2017-07-18-Transition-Proteins-for-REVIGO.csv), I sorted through the list and picked the lowest p-value for each protein (all 20 replicates were listed) and only kept the first GOterm listed in the original \"goterm\" column. The resultant file can be found [here](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-08-Final-Transitions/2017-07-18-Transition-Proteins-for-REVIGO-UNIQUE.csv).\n",
"\n",
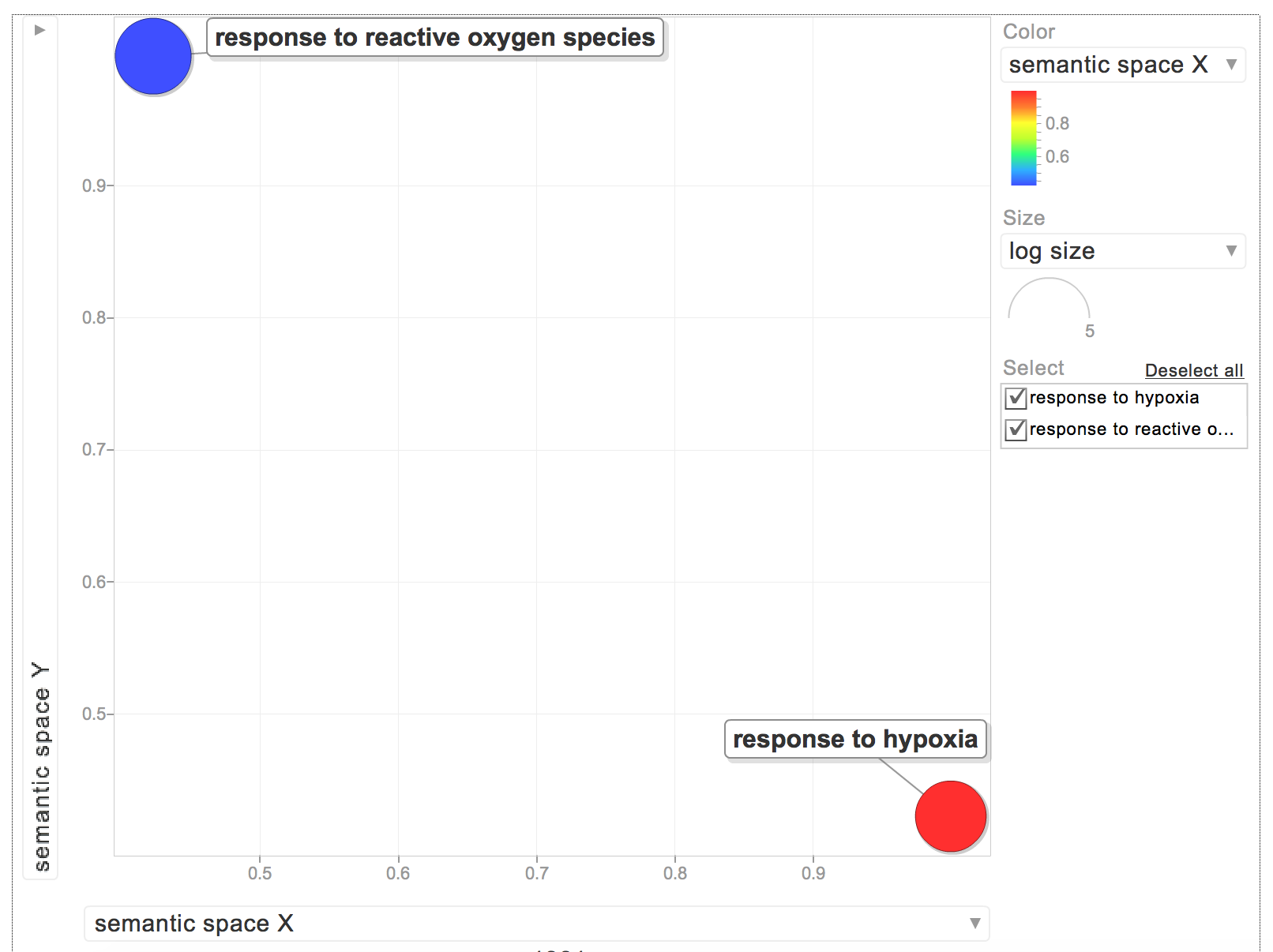
"When I put the GOterms and p-values into REVIGO, I only had 2 biological process GOterms:\n",
"\n",
"\n",
"\n",
"The rest were for molecular functions:\n",
"\n",
"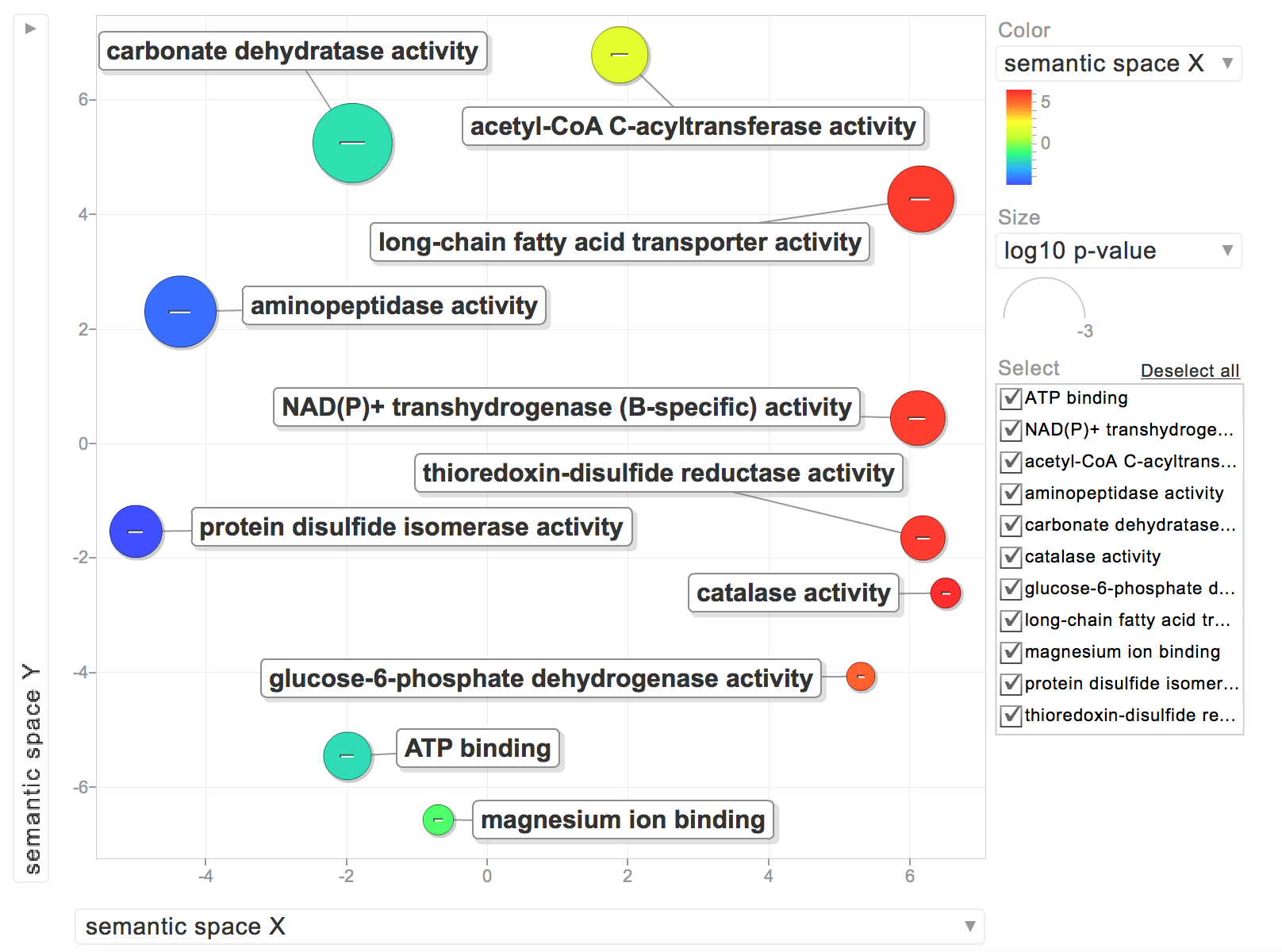\n",
"\n",
"While interesting, I'd rather have all of my GOterms be relevant to a biological process. I need to find an efficient way to sort through my list of GOterms and see if they are for biological processes."
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"**Update 4 p.m.**: I posted [this Github issue](https://github.com/sr320/LabDocs/issues/660#issuecomment-316159820) to figure it out. Based on Steven's comments, I'm going to forgo any REVIGO visualization since it may not be the most appropriate. I'll stick with my heatmap for now!"
]
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
}
],
"metadata": {
"anaconda-cloud": {},
"kernelspec": {
"display_name": "Python [default]",
"language": "python",
"name": "python3"
},
"language_info": {
"codemirror_mode": {
"name": "ipython",
"version": 3
},
"file_extension": ".py",
"mimetype": "text/x-python",
"name": "python",
"nbconvert_exporter": "python",
"pygments_lexer": "ipython3",
"version": "3.5.2"
}
},
"nbformat": 4,
"nbformat_minor": 1
}
"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Final Transition List\n",
"\n",
"[Final transitions](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-08-Final-Transitions/2017-07-09-Final-SRM-Transitions.csv)\n",
"\n",
"[Final Skyline document](http://owl.fish.washington.edu/spartina/DNR_Skyline_SRM_20170707/Gigas-7-9-Final-SRM-Transitions.sky.zip)"
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"## Update 2017-07-10"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Emma forgot that the 150 transition target maximum needs to include PRTC peptides! There are 38 transitions from PRTC peptides that we added, so we needed to cut down on my transition targets.\n",
"\n",
"The first thing we did was look at the final target list:\n",
"\n",
"\n",
"\n",
"In this list, there are proteins that have more than three target transitions per peptide. For the peptides with more than three associated transitions, we removed the least abundant ones so that we had a total of three transitions at the end. Additionally, the two catalase proteins, CHOYPCATA1.3|m.1120 and CHOYPCATA3.3|m.21642, had two peptides associated with both of them, LVENIGNHLINTQK and LQAHLDSVSNVSK. We deleted the redundant listing of these peptides.\n",
"\n",
"After this, we still needed to reduce the number of transitions, so I referred to the notes I had from my final review:\n",
"\n",
"\n",
"\n",
"There were two proteins that had one slightly sloppy peptide each, so we deleted them. Our [revised final transition list](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-08-Final-Transitions/2017-07-10-SRM-Transitions-With-PRTC.csv) has 159 total transitions. The 150 transition maximum is there because the machine can reliably detect 150 transitions at a time. We're pushing it with 159, so we will check our data to ensure our method is good. I also [revised my Skyline document](http://owl.fish.washington.edu/spartina/DNR_Skyline_SRM_20170707/2017-07-10-FINAL-SRM-Transitions-with-PRTC/Gigas-7-10-Final-Transition-List.sky.zip) to reflect these changes."
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"## Update 2017-07-11"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Predicted Retention Time Calculations\n",
"\n",
"Now that the [SRM assay is running](https://yaaminiv.github.io/SRM-Assay-Day1/), I need to confirm that I am getting the data I want. To do this, Emma and I selected a few PRTC peptides to find retention times for:\n",
"\n",
"- LTILEELR\n",
"- ELGQSGVDTYLQTK\n",
"- SAAGAFGPELSR\n",
"- DIPVPKPK\n",
"- HVLTSIGEK\n",
"- SSAAPPPPPR\n",
"\n",
"Using the fifth SRM QC run, I got retention times for these peptides. Emma went through 2 oyster DIA files (oyster 5 and oyster 10) and one geoduck DIA file's data and found retention times for our previous mass spectrometer run. I then regressed the SRM retention times against the DIA retention times to get a linear equation.\n",
"\n",
"\n",
"\n",
"Using this equation and DIA retention times for my transition targets, I calculated predicted SRM retention times. My data can be found [here](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-08-Final-Transitions/2017-07-11-Predicted-SRM-Retention-Times.xlsx). "
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Retention Time Confirmation\n",
"\n",
"In Skyline, I went through each peptide in two of my newly-collected oyster samples to ensure that Skyline picked the right peak. While the peaks were present around their calculation retention time, Skyline did not pick that peak for all but a few peptides.\n",
"\n",
"For example, Skyline chose a peak at 34.4 min for this peptide, when the best peak is clearly around 28 minutes. The best peak matches the best retention time I calculated as well.\n",
"\n",
"\n",
"\n",
"To fix this, I clicked on the peak I wanted Skyline to measure. Once I clicked on it, a black arrow appeared next to it.\n",
"\n",
"\n",
"\n",
"I then right-clicked and selected \"Apply Peak to All\", which had Skyline select the best peak for all samples in the document. Below, you can see that both samples have peaks made of three coeluting fragments around 28 minutes.\n",
"\n",
"\n",
"\n",
"I went through this process for all of my peaks. While Skyline may not be fully capable of picking the best peaks, the peaks were present! This is confirmation that the SRM assay works."
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"## Update 2017-07-18"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Since I have some time before I need to analyze my SRM data, I wanted to find ways to visualize my final transition list. The past few times I made NMDS plots, heatmaps and REVIGO plots, so I figured I would do the same this time. I used code [in this R script](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-08-Final-Transitions/2017-07-18-Final-Transition-Visualizations.R)."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### NMDS plot\n",
"\n",
"\n",
"\n",
"I ended up with all but one site and eelgrass condition nested on top of eachother. Not entire sure waht this means. I'll need to consult Emma to ensure that I did this correctly."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Heatmap\n",
"\n",
"\n",
"\n",
"My heatmap is a much cleaner way to visualize the proteins I selected for my final transition list, and how the expression differs between sites."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### REVIGO\n",
"\n",
"After I exported a list of [proteins, GOterms and p-values](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-08-Final-Transitions/2017-07-18-Transition-Proteins-for-REVIGO.csv), I sorted through the list and picked the lowest p-value for each protein (all 20 replicates were listed) and only kept the first GOterm listed in the original \"goterm\" column. The resultant file can be found [here](https://github.com/RobertsLab/project-oyster-oa/blob/master/analyses/DNR_TransitionSelection_20170707/2017-07-08-Final-Transitions/2017-07-18-Transition-Proteins-for-REVIGO-UNIQUE.csv).\n",
"\n",
"When I put the GOterms and p-values into REVIGO, I only had 2 biological process GOterms:\n",
"\n",
"\n",
"\n",
"The rest were for molecular functions:\n",
"\n",
"\n",
"\n",
"While interesting, I'd rather have all of my GOterms be relevant to a biological process. I need to find an efficient way to sort through my list of GOterms and see if they are for biological processes."
]
},
{
"cell_type": "markdown",
"metadata": {
"collapsed": true
},
"source": [
"**Update 4 p.m.**: I posted [this Github issue](https://github.com/sr320/LabDocs/issues/660#issuecomment-316159820) to figure it out. Based on Steven's comments, I'm going to forgo any REVIGO visualization since it may not be the most appropriate. I'll stick with my heatmap for now!"
]
},
{
"cell_type": "code",
"execution_count": null,
"metadata": {
"collapsed": true
},
"outputs": [],
"source": []
}
],
"metadata": {
"anaconda-cloud": {},
"kernelspec": {
"display_name": "Python [default]",
"language": "python",
"name": "python3"
},
"language_info": {
"codemirror_mode": {
"name": "ipython",
"version": 3
},
"file_extension": ".py",
"mimetype": "text/x-python",
"name": "python",
"nbconvert_exporter": "python",
"pygments_lexer": "ipython3",
"version": "3.5.2"
}
},
"nbformat": 4,
"nbformat_minor": 1
}