06.2-cod-RNAseq-alignment-genome
================
Kathleen Durkin
2024-04-16
- 1 Create a Bash variables
file
- 2 Align to reference genome
(Hisat2)
- 2.1 Retrieving the
reference genome and gff
- 2.2 Verify genome FastA MD5
checksum
- 2.3 Building
Index
- 2.4 Alignment
- 3 Read
Summarization
- 3.1 Exons
- 3.2 Genes
``` r
library(tidyverse)
library(dplyr)
library(magrittr)
library(knitr)
library(ggplot2)
library(plotly)
```
Code for aligning RNAseq data to reference genome, to be used on
[Pacific cod RNAseq
data](https://shedurkin.github.io/Roberts-LabNotebook/posts/projects/pacific_cod/2023_12_13_pacific_cod.html).
- Raw reads found
[here](https://owl.fish.washington.edu/nightingales/G_macrocephalus/30-943133806/)
- Trimmed reads:
- Genome downloaded from
[NCBI](https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_031168955.1/),
stored
[here](https://owl.fish.washington.edu/halfshell/genomic-databank/GCF_031168955.1_ASM3116895v1_rna.fna)
as a part of lab [genomic
resources](https://robertslab.github.io/resources/Genomic-Resources/#gadus-macrocephalus-pacific-cod)
- Genome GTF downloaded from
[NCBI](https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_031168955.1/)
# 1 Create a Bash variables file
This allows usage of Bash variables (e.g. paths to common directories)
across R Markdown chunks.
``` bash
{
echo "#### Assign Variables ####"
echo ""
echo "# Data directories"
echo 'export cod_dir=/home/shared/8TB_HDD_02/shedurkin/project-cod-temperature'
echo 'export output_dir_top=${cod_dir}/output/06.2-cod-RNAseq-alignment-genome'
echo 'export genome_fasta_dir=${cod_dir}/data'
echo 'export trimmed_reads_dir=${cod_dir}/output/05-cod-RNAseq-trimming/trimmed-reads'
echo ""
echo "# Input/Output files"
echo 'export genome_fasta_name="GCF_031168955.1_ASM3116895v1_genomic"'
echo 'export genome_fasta="${genome_fasta_dir}/${genome_fasta_name}"'
echo 'export genome_gtf_name="genomic"'
echo 'export genome_gtf="${genome_fasta_dir}/${genome_gtf_name}"'
echo 'export hisat2_exon_name="G_macrocephalus_exon"'
echo 'export hisat2_exon="${hisat2_output_dir}/${hisat_exon_name}"'
echo 'export hisat2_splice_sites_name="G_macrocephalus_splice_sites"'
echo 'export hisat2_splice_sites="${hisat2_output_dir}/${hisat2_splice_sites_name}"'
echo 'export hisat2_index_name="G_macrocephalus_Hisat2_index"'
echo 'export hisat2_index="${hisat2_output_dir}/${hisat2_index}"'
echo "# External data URLs and checksums"
echo 'export genome_fasta_url="https://owl.fish.washington.edu/halfshell/genomic-databank/GCF_031168955.1_ASM3116895v1_genomic.fna"'
echo 'export genome_fasta_checksum="5144890d4eceb0b258d92db3f35c681e"'
echo 'export genome_gtf_url="https://api.ncbi.nlm.nih.gov/datasets/v2alpha/genome/accession/GCF_031168955.1/download?include_annotation_type=GENOME_GTF"'
#echo 'export genome_gtf_checksum="173fb3c159e474391c5c4aa1f7230024"'
echo "# Paths to programs"
echo 'export hisat2_exons=/home/shared/hisat2-2.2.1/hisat2_extract_exons.py'
echo 'export hisat2_splice_sites=/home/shared/hisat2-2.2.1/hisat2_extract_splice_sites.py'
echo 'export hisat2_build=/home/shared/hisat2-2.2.1/hisat2-build'
echo ""
echo "# Set number of CPUs to use"
echo 'export threads=20'
echo ""
echo "# Programs associative array"
echo "declare -A programs_array"
echo "programs_array=("
echo '[hisat2_exons]="${hisat2_exons}" \'
echo '[hisat2_splice_sites]="${hisat2_splice_sites}" \'
echo '[hisat2_build]="${hisat2_build}" \'
echo '[trinity_abund_to_matrix]="${trinity_abund_to_matrix}" \'
echo ")"
} > .bashvars
cat .bashvars
```
#### Assign Variables ####
# Data directories
export cod_dir=/home/shared/8TB_HDD_02/shedurkin/project-cod-temperature
export output_dir_top=${cod_dir}/output/06.2-cod-RNAseq-alignment-genome
export genome_fasta_dir=${cod_dir}/data
export trimmed_reads_dir=${cod_dir}/output/05-cod-RNAseq-trimming/trimmed-reads
# Input/Output files
export genome_fasta_name="GCF_031168955.1_ASM3116895v1_genomic"
export genome_fasta="${genome_fasta_dir}/${genome_fasta_name}"
export genome_gtf_name="genomic"
export genome_gtf="${genome_fasta_dir}/${genome_gtf_name}"
export hisat2_exon_name="G_macrocephalus_exon"
export hisat2_exon="${hisat2_output_dir}/${hisat_exon_name}"
export hisat2_splice_sites_name="G_macrocephalus_splice_sites"
export hisat2_splice_sites="${hisat2_output_dir}/${hisat2_splice_sites_name}"
export hisat2_index_name="G_macrocephalus_Hisat2_index"
export hisat2_index="${hisat2_output_dir}/${hisat2_index}"
# External data URLs and checksums
export genome_fasta_url="https://owl.fish.washington.edu/halfshell/genomic-databank/GCF_031168955.1_ASM3116895v1_genomic.fna"
export genome_fasta_checksum="5144890d4eceb0b258d92db3f35c681e"
export genome_gtf_url="https://api.ncbi.nlm.nih.gov/datasets/v2alpha/genome/accession/GCF_031168955.1/download?include_annotation_type=GENOME_GTF"
# Paths to programs
export hisat2_exons=/home/shared/hisat2-2.2.1/hisat2_extract_exons.py
export hisat2_splice_sites=/home/shared/hisat2-2.2.1/hisat2_extract_splice_sites.py
export hisat2_build=/home/shared/hisat2-2.2.1/hisat2-build
# Set number of CPUs to use
export threads=20
# Programs associative array
declare -A programs_array
programs_array=(
[hisat2_exons]="${hisat2_exons}" \
[hisat2_splice_sites]="${hisat2_splice_sites}" \
[hisat2_build]="${hisat2_build}" \
[trinity_abund_to_matrix]="${trinity_abund_to_matrix}" \
)
# 2 Align to reference genome (Hisat2)
## 2.1 Retrieving the reference genome and gff
``` bash
# Load bash variables into memory
source .bashvars
wget \
--directory-prefix ${genome_fasta_dir} \
--recursive \
--no-check-certificate \
--continue \
--no-host-directories \
--no-directories \
--no-parent \
--quiet \
--execute robots=off \
--accept "${genome_fasta_name}.fna" ${genome_fasta_url}
```
v NOT CURRENTLY WORKING v, had to download locally and then upload to
server for use
``` bash
# Load bash variables into memory
source .bashvars
cd ../data
curl -O "https://api.ncbi.nlm.nih.gov/datasets/v2alpha/genome/accession/GCF_031168955.1/download?include_annotation_type=GENOME_GTF"
#curl -O "${genome_gtf_url}"
```
``` bash
# Load bash variables into memory
source .bashvars
ls -lh "${genome_fasta_dir}"
```
total 1.9G
drwxr-xr-x 3 shedurkin labmembers 4.0K Mar 4 11:05 05-cod-RNAseq-trimming
-rw-r--r-- 1 shedurkin labmembers 13K Dec 27 15:45 Cod_RNAseq_NGS_Template_File.xlsx
-rw-r--r-- 1 shedurkin labmembers 2.1K Mar 20 20:55 DESeq2_Sample_Information.csv
-rw-r--r-- 1 shedurkin labmembers 38M Oct 25 2023 Gadus_macrocephalus.coding.gene.V1.cds
-rw-r--r-- 1 shedurkin labmembers 537M Oct 16 2023 GCF_031168955.1_ASM3116895v1_genomic.fna
-rw-r--r-- 1 shedurkin labmembers 351M Oct 16 2023 GCF_031168955.1_ASM3116895v1.gff
-rw-r--r-- 1 shedurkin labmembers 169M Oct 16 2023 GCF_031168955.1_ASM3116895v1_rna.fna
-rw-r--r-- 1 shedurkin labmembers 404M Apr 23 14:29 genomic.gtf
-rw-r--r-- 1 shedurkin labmembers 47K Oct 25 2023 Pcod Temp Growth experiment 2022-23 DATA.xlsx
-rw-r--r-- 1 shedurkin labmembers 231K Mar 4 17:41 Sample.QC.report.of_30-943133806_240118025106.pdf
-rw-r--r-- 1 shedurkin labmembers 12K Mar 4 17:41 Sample.QC.report.of_30-943133806_240118025106.xlsx
-rw-r--r-- 1 shedurkin labmembers 12K Oct 25 2023 temp-experiment.csv
-rw-r--r-- 1 shedurkin labmembers 271M Oct 25 2023 uniprot_sprot_r2023_04.fasta
-rw-r--r-- 1 shedurkin labmembers 88M Apr 17 11:54 uniprot_sprot_r2023_04.fasta.gz
## 2.2 Verify genome FastA MD5 checksum
``` bash
# Load bash variables into memory
source .bashvars
cd "${genome_fasta_dir}"
# Checksums file contains other files, so this just looks for the sRNAseq files.
md5sum --check <<< "${genome_fasta_checksum} ${genome_fasta_name}.fna"
```
``` bash
# Load bash variables into memory
source .bashvars
cd "${genome_fasta_dir}"
# Checksums file contains other files, so this just looks for the sRNAseq files.
md5sum --check <<< "${genome_gtf_checksum} ${genome_gtf_name}.gtf"
```
## 2.3 Building Index
``` bash
# Load bash variables into memory
source .bashvars
# Create Hisat2 exons tab file
/home/shared/hisat2-2.2.1/hisat2_extract_exons.py \
../data/genomic.gtf \
> ../output/06.2-cod-RNAseq-alignment-genome/hisat2/G_macrocephalus_exon.tab
# Create Hisat2 exons tab file
#"${programs_array[hisat2_exons]}" \
#"${genome_gtf}.gtf" \
#> "${hisat2_exon}.tab"
```
``` bash
# Create Hisat2 splice sites tab file
/home/shared/hisat2-2.2.1/hisat2_extract_splice_sites.py \
../data/genomic.gtf \
> ../output/06.2-cod-RNAseq-alignment-genome/hisat2/G_macrocephalus_splice_sites.tab
#"${programs_array[hisat2_splice_sites]}" \
#"${genome_gtf}.gtf" \
#> "${hisat2_splice_sites}.tab"
```
``` bash
# Build Hisat2 reference index using splice sites and exons
/home/shared/hisat2-2.2.1/hisat2-build \
../data/GCF_031168955.1_ASM3116895v1_genomic.fna \
../output/06.2-cod-RNAseq-alignment-genome/hisat2/G_macrocephalus_index.idx \
--exon ../output/06.2-cod-RNAseq-alignment-genome/hisat2/G_macrocephalus_exon.tab \
--ss ../output/06.2-cod-RNAseq-alignment-genome/hisat2/G_macrocephalus_splice_sites.tab \
-p 20 \
2> ../output/06.2-cod-RNAseq-alignment-genome/hisat2/hisat2-build_stats.txt
#"${programs_array[hisat2_build]}" \
#"${genome_fasta}.fna" \
#"${hisat2_index}.idx" \
#--exon "${hisat2_exon}.tab" \
#--ss "${hisat2_splice_sites}.tab" \
#-p "${threads}" \
#2> "${output_dir_top}/hisat2-build_stats.txt"
```
``` bash
# Load bash variables into memory
source .bashvars
ls -lh "${output_dir_top}"
```
total 56K
drwxr-xr-x 3 shedurkin labmembers 4.0K Apr 30 15:04 featureCounts-exon
drwxr-xr-x 3 shedurkin labmembers 36K May 1 15:34 featureCounts-gene
drwxr-xr-x 2 shedurkin labmembers 12K Apr 30 14:23 hisat2
## 2.4 Alignment
``` bash
# Load bash variables into memory
source .bashvars
## Sample Quantification
# Hisat2 alignments
find ../output/05-cod-RNAseq-trimming/trimmed-reads/*.gz \
| xargs basename -s .flexbar_trim.R_1.fastq.gz | xargs -I{} \
/home/shared/hisat2-2.2.1/hisat2 \
-x ../output/06.2-cod-RNAseq-alignment-genome/hisat2/G_macrocephalus_index.idx \
-p 20 \
-1 ../output/05-cod-RNAseq-trimming/trimmed-reads/{}.flexbar_trim.R_1.fastq.gz \
-2 ../output/05-cod-RNAseq-trimming/trimmed-reads/{}.flexbar_trim.R_2.fastq.gz \
-S ../output/06.2-cod-RNAseq-alignment-genome/hisat2/{}.sam
&> ../output/06.2-cod-RNAseq-alignment-genome/hisat2/{}_hisat2.log
# # Hisat2 alignments
# find ${trimmed_reads_dir}/*.gz \
# | xargs basename -s .flexbar_trim.R_1.fastq.gz | xargs -I{} \
# "${programs_array[hisat2]}" \
# -x "${hisat2_index}.idx" \
# -p 20 \
# -1 ${trimmed_reads_dir}/{}.flexbar_trim.R_1.fastq.gz \
# -2 ${trimmed_reads_dir}/{}.flexbar_trim.R_2.fastq.gz \
# -S ${output_dir_top}/{}.sam
#
```
``` bash
# Sort SAM files, convert to BAM, and index
for samfile in ../output/06.2-cod-RNAseq-alignment-genome/hisat2/*.sam; do
bamfile="${samfile%.sam}.bam"
sorted_bamfile="${samfile%.sam}.sorted.bam"
# Check if the output file already exists
if [[ ! -e "$sorted_bamfile" ]]; then
# Convert SAM to BAM
/home/shared/samtools-1.12/samtools view -bS -@ 10 "$samfile" > "$bamfile"
# Sort BAM
/home/shared/samtools-1.12/samtools sort -@ 10 "$bamfile" -o "$sorted_bamfile"
# Index sorted BAM
/home/shared/samtools-1.12/samtools index -@ 10 "$sorted_bamfile"
fi
done
```
``` bash
# Count the number of samples for which we have sorted bam files -- I should have 79 (one for each input sample)
find ../output/06.2-cod-RNAseq-alignment-genome/hisat2/ -type f -name "*.sorted.bam" | wc -l
```
79
``` bash
# Delete unneccessary index files
rm ../output/06.2-cod-RNAseq-alignment-genome/hisat2/*.ht2
# Delete unneeded SAM files
rm ../output/06.2-cod-RNAseq-alignment-genome/hisat2/*.sam
# # Sort SAM files, convert to BAM, and index
# ${programs_array[samtools_view]} \
# -@ "${threads}" \
# -Su "${sample_name}".sam \
# | ${programs_array[samtools_sort]} - \
# -@ "${threads}" \
# -o "${sample_name}".sorted.bam
# ${programs_array[samtools_index]} "${sample_name}".sorted.bam
#
#
# # Delete unneccessary index files
# rm "${genome_index_name}"*.ht2
#
# # Delete unneeded SAM files
# rm ./*.sam
```
``` bash
# View the output files
ls -lh ../output/06.2-cod-RNAseq-alignment-genome/hisat2/
```
total 364G
-rw-r--r-- 1 shedurkin labmembers 2.7G Apr 24 12:27 100.bam
-rw-r--r-- 1 shedurkin labmembers 1.6G Apr 24 12:28 100.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.1M Apr 24 12:28 100.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.6G Apr 24 12:29 107.bam
-rw-r--r-- 1 shedurkin labmembers 1.5G Apr 24 12:30 107.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 852K Apr 24 12:30 107.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.3G Apr 24 12:32 108.bam
-rw-r--r-- 1 shedurkin labmembers 2.1G Apr 24 12:35 108.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.3M Apr 24 12:35 108.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.4G Apr 24 12:39 109.bam
-rw-r--r-- 1 shedurkin labmembers 2.0G Apr 24 12:41 109.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 24 12:41 109.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.9G Apr 24 12:48 10.bam
-rw-r--r-- 1 shedurkin labmembers 1.8G Apr 24 12:49 10.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 24 12:49 10.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.2G Apr 24 12:58 110.bam
-rw-r--r-- 1 shedurkin labmembers 1.9G Apr 24 13:00 110.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.1M Apr 24 13:01 110.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.3G Apr 24 13:09 117.bam
-rw-r--r-- 1 shedurkin labmembers 2.0G Apr 24 13:12 117.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.3M Apr 24 13:12 117.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.3G Apr 24 13:21 118.bam
-rw-r--r-- 1 shedurkin labmembers 2.0G Apr 24 13:24 118.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.3M Apr 24 13:24 118.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.4G Apr 24 13:27 119.bam
-rw-r--r-- 1 shedurkin labmembers 2.0G Apr 24 13:30 119.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.1M Apr 24 13:30 119.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.3G Apr 24 13:33 11.bam
-rw-r--r-- 1 shedurkin labmembers 2.0G Apr 24 13:35 11.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 24 13:35 11.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.1G Apr 24 13:38 120.bam
-rw-r--r-- 1 shedurkin labmembers 1.9G Apr 24 13:40 120.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 24 13:40 120.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.2G Apr 24 13:43 121.bam
-rw-r--r-- 1 shedurkin labmembers 2.0G Apr 24 13:45 121.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 816K Apr 24 13:45 121.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.6G Apr 24 13:53 127.bam
-rw-r--r-- 1 shedurkin labmembers 2.2G Apr 24 13:55 127.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 24 13:55 127.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 1.6G Apr 24 13:57 128.bam
-rw-r--r-- 1 shedurkin labmembers 940M Apr 24 13:58 128.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 713K Apr 24 13:58 128.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 1.4G Apr 24 14:01 129.bam
-rw-r--r-- 1 shedurkin labmembers 1.1G Apr 24 14:02 129.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.9M Apr 24 14:02 129.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.1G Apr 24 14:07 12.bam
-rw-r--r-- 1 shedurkin labmembers 1.9G Apr 24 14:09 12.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 24 14:09 12.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 1.4G Apr 24 14:12 131.bam
-rw-r--r-- 1 shedurkin labmembers 883M Apr 24 14:12 131.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 736K Apr 24 14:12 131.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 1.3G Apr 24 14:15 137.bam
-rw-r--r-- 1 shedurkin labmembers 780M Apr 24 14:15 137.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 659K Apr 24 14:15 137.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 1.3G Apr 24 14:16 138.bam
-rw-r--r-- 1 shedurkin labmembers 773M Apr 24 14:17 138.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 656K Apr 24 14:17 138.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 1.5G Apr 24 14:18 139.bam
-rw-r--r-- 1 shedurkin labmembers 906M Apr 24 14:18 139.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 786K Apr 24 14:19 139.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.8G Apr 24 14:20 13.bam
-rw-r--r-- 1 shedurkin labmembers 1.7G Apr 24 14:22 13.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 24 14:22 13.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 1.5G Apr 24 14:23 140.bam
-rw-r--r-- 1 shedurkin labmembers 904M Apr 24 14:23 140.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 749K Apr 24 14:23 140.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 1.3G Apr 24 14:24 147.bam
-rw-r--r-- 1 shedurkin labmembers 812M Apr 24 14:25 147.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 692K Apr 24 14:25 147.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.6G Apr 24 14:27 148.bam
-rw-r--r-- 1 shedurkin labmembers 2.2G Apr 24 14:30 148.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.1M Apr 24 14:30 148.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 15G Apr 24 14:43 149.bam
-rw-r--r-- 1 shedurkin labmembers 8.9G Apr 24 14:54 149.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 7.1M Apr 24 14:55 149.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.8G Apr 24 14:58 150.bam
-rw-r--r-- 1 shedurkin labmembers 2.4G Apr 24 15:00 150.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.3M Apr 24 15:00 150.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.7G Apr 24 15:03 18.bam
-rw-r--r-- 1 shedurkin labmembers 1.6G Apr 24 15:05 18.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 24 15:05 18.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.7G Apr 24 15:16 19.bam
-rw-r--r-- 1 shedurkin labmembers 3.5G Apr 24 15:09 19-G.bam
-rw-r--r-- 1 shedurkin labmembers 2.3G Apr 24 15:12 19-G.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.1M Apr 24 15:12 19-G.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.7G Apr 24 15:21 19-S.bam
-rw-r--r-- 1 shedurkin labmembers 1.6G Apr 24 15:17 19.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1023K Apr 24 15:17 19.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.4G Apr 24 15:24 19-S.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 24 15:25 19-S.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.2G Apr 24 15:27 1.bam
-rw-r--r-- 1 shedurkin labmembers 1.9G Apr 24 15:29 1.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.1M Apr 24 15:29 1.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.6G Apr 24 15:37 20.bam
-rw-r--r-- 1 shedurkin labmembers 3.6G Apr 24 15:32 20-G.bam
-rw-r--r-- 1 shedurkin labmembers 2.4G Apr 24 15:34 20-G.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.1M Apr 24 15:35 20-G.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.5G Apr 24 15:40 20-S.bam
-rw-r--r-- 1 shedurkin labmembers 1.6G Apr 24 15:38 20.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.1M Apr 24 15:38 20.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 1.7G Apr 24 15:41 20-S.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 935K Apr 24 15:41 20-S.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.7G Apr 24 15:43 21.bam
-rw-r--r-- 1 shedurkin labmembers 1.6G Apr 24 15:44 21.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 24 15:44 21.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.8G Apr 24 15:46 28.bam
-rw-r--r-- 1 shedurkin labmembers 1.7G Apr 24 15:47 28.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.1M Apr 24 15:47 28.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.8G Apr 24 15:49 29.bam
-rw-r--r-- 1 shedurkin labmembers 1.8G Apr 24 15:51 29.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 24 15:51 29.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.0G Apr 24 15:53 2.bam
-rw-r--r-- 1 shedurkin labmembers 1.8G Apr 24 15:54 2.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 990K Apr 24 15:54 2.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.0G Apr 24 15:56 30.bam
-rw-r--r-- 1 shedurkin labmembers 1.8G Apr 24 15:58 30.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.1M Apr 24 15:58 30.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.0G Apr 24 16:00 31.bam
-rw-r--r-- 1 shedurkin labmembers 1.7G Apr 24 16:01 31.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 839K Apr 24 16:01 31.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.4G Apr 24 16:03 37.bam
-rw-r--r-- 1 shedurkin labmembers 1.4G Apr 24 16:04 37.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 847K Apr 24 16:04 37.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.7G Apr 24 16:06 38.bam
-rw-r--r-- 1 shedurkin labmembers 1.7G Apr 24 16:08 38.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.3M Apr 24 16:08 38.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.7G Apr 24 16:10 39.bam
-rw-r--r-- 1 shedurkin labmembers 1.6G Apr 24 16:11 39.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 936K Apr 24 16:11 39.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.0G Apr 24 16:13 3.bam
-rw-r--r-- 1 shedurkin labmembers 1.9G Apr 24 16:15 3.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.3M Apr 24 16:15 3.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.4G Apr 24 16:17 40.bam
-rw-r--r-- 1 shedurkin labmembers 1.5G Apr 24 16:18 40.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.1M Apr 24 16:18 40.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.4G Apr 24 16:20 41.bam
-rw-r--r-- 1 shedurkin labmembers 1.6G Apr 24 16:22 41.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.3M Apr 24 16:22 41.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.7G Apr 26 16:47 47.bam
-rw-r--r-- 1 shedurkin labmembers 1.7G Apr 26 16:49 47.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 989K Apr 26 16:49 47.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.9G Apr 26 16:51 48.bam
-rw-r--r-- 1 shedurkin labmembers 1.7G Apr 26 16:53 48.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 927K Apr 26 16:53 48.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.0G Apr 26 16:55 49.bam
-rw-r--r-- 1 shedurkin labmembers 1.8G Apr 26 16:57 49.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.1M Apr 26 16:57 49.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.3G Apr 26 16:59 4.bam
-rw-r--r-- 1 shedurkin labmembers 1.9G Apr 26 17:02 4.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.1M Apr 26 17:02 4.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.9G Apr 26 17:04 50.bam
-rw-r--r-- 1 shedurkin labmembers 1.7G Apr 26 17:05 50.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.1M Apr 26 17:05 50.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.9G Apr 26 17:14 57.bam
-rw-r--r-- 1 shedurkin labmembers 4.3G Apr 26 17:09 57-G.bam
-rw-r--r-- 1 shedurkin labmembers 2.8G Apr 26 17:11 57-G.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 26 17:11 57-G.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 1.8G Apr 26 17:17 57-S.bam
-rw-r--r-- 1 shedurkin labmembers 1.8G Apr 26 17:15 57.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.1M Apr 26 17:15 57.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 1.4G Apr 26 17:18 57-S.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1001K Apr 26 17:18 57-S.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.9G Apr 26 17:27 58.bam
-rw-r--r-- 1 shedurkin labmembers 4.4G Apr 26 17:22 58-G.bam
-rw-r--r-- 1 shedurkin labmembers 2.8G Apr 26 17:24 58-G.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.3M Apr 26 17:25 58-G.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.2G Apr 26 17:30 58-S.bam
-rw-r--r-- 1 shedurkin labmembers 1.8G Apr 26 17:28 58.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.1M Apr 26 17:29 58.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 1.4G Apr 26 17:32 58-S.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 745K Apr 26 17:32 58-S.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.9G Apr 26 17:34 59.bam
-rw-r--r-- 1 shedurkin labmembers 1.7G Apr 26 17:36 59.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 899K Apr 26 17:36 59.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.0G Apr 26 17:38 5.bam
-rw-r--r-- 1 shedurkin labmembers 1.8G Apr 26 17:40 5.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.3M Apr 26 17:40 5.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.1G Apr 26 17:43 60.bam
-rw-r--r-- 1 shedurkin labmembers 1.9G Apr 26 17:45 60.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 26 17:45 60.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.2G Apr 26 17:47 67.bam
-rw-r--r-- 1 shedurkin labmembers 1.9G Apr 26 17:49 67.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 26 17:49 67.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.1G Apr 26 17:52 68.bam
-rw-r--r-- 1 shedurkin labmembers 1.9G Apr 26 17:54 68.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 26 17:54 68.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.6G Apr 26 17:56 69.bam
-rw-r--r-- 1 shedurkin labmembers 1.6G Apr 26 17:58 69.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 944K Apr 26 17:58 69.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.2G Apr 26 17:59 70.bam
-rw-r--r-- 1 shedurkin labmembers 1.4G Apr 26 18:01 70.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 914K Apr 26 18:01 70.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.6G Apr 26 18:03 78.bam
-rw-r--r-- 1 shedurkin labmembers 1.6G Apr 26 18:04 78.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 26 18:05 78.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.8G Apr 26 18:07 79.bam
-rw-r--r-- 1 shedurkin labmembers 1.7G Apr 26 18:08 79.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.1M Apr 26 18:09 79.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.2G Apr 26 18:10 80.bam
-rw-r--r-- 1 shedurkin labmembers 1.4G Apr 26 18:12 80.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.1M Apr 26 18:12 80.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.4G Apr 26 18:14 83.bam
-rw-r--r-- 1 shedurkin labmembers 1.4G Apr 26 18:15 83.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 967K Apr 26 18:15 83.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.8G Apr 26 18:17 88.bam
-rw-r--r-- 1 shedurkin labmembers 1.8G Apr 26 18:19 88.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 26 18:19 88.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.6G Apr 26 18:21 90.bam
-rw-r--r-- 1 shedurkin labmembers 1.6G Apr 26 18:23 90.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 26 18:23 90.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.0G Apr 26 18:25 91.bam
-rw-r--r-- 1 shedurkin labmembers 1.9G Apr 26 18:27 91.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 26 18:27 91.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.5G Apr 26 18:29 97.bam
-rw-r--r-- 1 shedurkin labmembers 1.5G Apr 26 18:31 97.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 943K Apr 26 18:31 97.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 3.3G Apr 26 18:34 98.bam
-rw-r--r-- 1 shedurkin labmembers 2.0G Apr 26 18:36 98.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.5M Apr 26 18:36 98.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.5G Apr 26 18:38 99.bam
-rw-r--r-- 1 shedurkin labmembers 1.6G Apr 26 18:40 99.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.2M Apr 26 18:40 99.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 7.4M Apr 23 14:34 G_macrocephalus_exon.tab
-rw-r--r-- 1 shedurkin labmembers 7.1M Apr 23 14:40 G_macrocephalus_splice_sites.tab
-rw-r--r-- 1 shedurkin labmembers 12K Apr 23 14:54 hisat2-build_stats.txt
-rw-r--r-- 1 shedurkin labmembers 2.4G Apr 26 18:42 RESUB-116.bam
-rw-r--r-- 1 shedurkin labmembers 1.5G Apr 26 18:43 RESUB-116.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1.1M Apr 26 18:43 RESUB-116.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.5G Apr 26 18:45 RESUB-156.bam
-rw-r--r-- 1 shedurkin labmembers 1.5G Apr 26 18:47 RESUB-156.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 974K Apr 26 18:47 RESUB-156.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.3G Apr 26 18:48 RESUB-36.bam
-rw-r--r-- 1 shedurkin labmembers 1.4G Apr 26 18:50 RESUB-36.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 852K Apr 26 18:50 RESUB-36.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.2G Apr 26 18:52 RESUB-76.bam
-rw-r--r-- 1 shedurkin labmembers 1.3G Apr 26 18:53 RESUB-76.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 895K Apr 26 18:53 RESUB-76.sorted.bam.bai
-rw-r--r-- 1 shedurkin labmembers 2.6G Apr 26 18:55 RESUB-94.bam
-rw-r--r-- 1 shedurkin labmembers 1.6G Apr 26 18:57 RESUB-94.sorted.bam
-rw-r--r-- 1 shedurkin labmembers 1014K Apr 26 18:57 RESUB-94.sorted.bam.bai
# 3 Read Summarization
Will be summarizing reads using
[featureCounts](https://www.rdocumentation.org/packages/Rsubread/versions/1.22.2/topics/featureCounts)
in the
[Rsubread](https://bioconductor.org/packages/release/bioc/vignettes/Rsubread/inst/doc/SubreadUsersGuide.pdf)
Bioconductor package
## 3.1 Exons
``` bash
/home/shared/subread-2.0.5-Linux-x86_64/bin/featureCounts \
-p --countReadPairs \
-T 5 \
-t exon \
-g gene_id \
-a ../data/genomic.gtf \
-o ../output/06.2-cod-RNAseq-alignment-genome/featureCounts-exon/featureCounts_exon_matrix.txt \
../output/06.2-cod-RNAseq-alignment-genome/hisat2/*.sorted.bam \
&> ../output/06.2-cod-RNAseq-alignment-genome/featureCounts-exon/featureCounts_exon.log
```
Save a version of the featureCounts count file with no header (aka
remove line 1, which contains the program and command info)
``` bash
sed '1d' ../output/06.2-cod-RNAseq-alignment-genome/featureCounts-exon/featureCounts_exon_matrix.txt > ../output/06.2-cod-RNAseq-alignment-genome/featureCounts-exon/featureCounts_exon_matrix_noheader.txt
```
``` bash
/home/sam/programs/mambaforge/bin/multiqc \
../output/06.2-cod-RNAseq-alignment-genome/featureCounts-exon/featureCounts_exon_matrix.txt.summary \
-o ../output/06.2-cod-RNAseq-alignment-genome/featureCounts-exon
# View directory contents
ls -lh ../output/06.2-cod-RNAseq-alignment-genome/featureCounts-exon
```
I also want to include the treatment/tank info when plotting alignment
rates across samples
``` r
# Load multiqc stats
featureCounts_exon_multiqc <- read.csv("../output/06.2-cod-RNAseq-alignment-genome/featureCounts-exon/multiqc_data/multiqc_featureCounts.txt", sep = '\t')
# Adjust sample name formatting (to prep for join)
featureCounts_exon_multiqc$Sample <- paste("sample_", featureCounts_exon_multiqc$Sample, sep = "")
# Load experimental data
cod_sample_info_OG <- read.csv("../data/DESeq2_Sample_Information.csv")
featureCounts_exon_multiqc_plustreatment <- left_join(cod_sample_info_OG, featureCounts_exon_multiqc, by = c("sample_name" = "Sample")) %>%
na.omit()
featureCounts_exon_multiqc_plustreatment <- featureCounts_exon_multiqc_plustreatment[order(featureCounts_exon_multiqc_plustreatment$sample_number),]
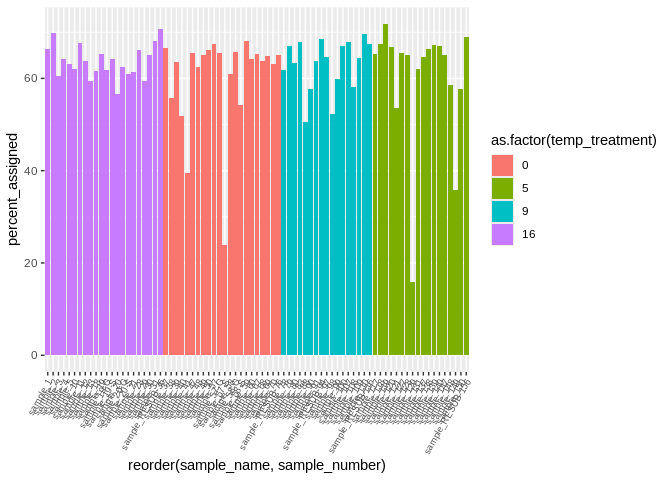
ggplot(featureCounts_exon_multiqc_plustreatment,
aes(x=reorder(sample_name, sample_number), y=percent_assigned, fill=as.factor(temp_treatment))) +
geom_bar(stat="identity") +
theme(axis.text.x = element_text(angle = 60, vjust = 1, hjust=1, size=7))
```
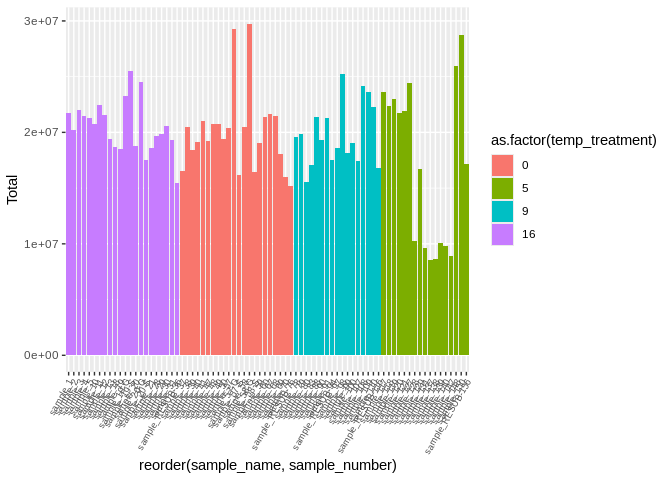
``` r
ggplot(featureCounts_exon_multiqc_plustreatment,
aes(x=reorder(sample_name, sample_number), y=Total, fill=as.factor(temp_treatment))) +
geom_bar(stat="identity") +
theme(axis.text.x = element_text(angle = 60, vjust = 1, hjust=1, size=7))
```
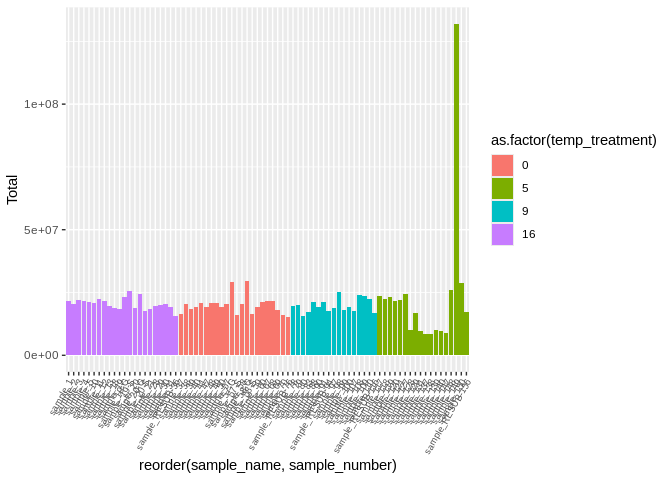
``` r
# sample149 is kind of throwing off the visualization, so lets remove and redo
ggplot(featureCounts_exon_multiqc_plustreatment[featureCounts_exon_multiqc_plustreatment$sample_name != "sample_149", ],
aes(x=reorder(sample_name, sample_number), y=Total, fill=as.factor(temp_treatment))) +
geom_bar(stat="identity") +
theme(axis.text.x = element_text(angle = 60, vjust = 1, hjust=1, size=7))
```
## 3.2 Genes
``` bash
/home/shared/subread-2.0.5-Linux-x86_64/bin/featureCounts \
-p --countReadPairs \
-T 5 \
-t gene \
-g gene_id \
-a ../data/genomic.gtf \
-o ../output/06.2-cod-RNAseq-alignment-genome/featureCounts-gene/featureCounts_gene_matrix.txt \
../output/06.2-cod-RNAseq-alignment-genome/hisat2/*.sorted.bam \
&> ../output/06.2-cod-RNAseq-alignment-genome/featureCounts-gene/featureCounts_gene.log
```
Save a version of the featureCounts count file with no header (aka
remove line 1, which contains the program and command info)
``` bash
sed '1d' ../output/06.2-cod-RNAseq-alignment-genome/featureCounts-gene/featureCounts_gene_matrix.txt > ../output/06.2-cod-RNAseq-alignment-genome/featureCounts-gene/featureCounts_gene_matrix_noheader.txt
```
``` bash
/home/sam/programs/mambaforge/bin/multiqc \
../output/06.2-cod-RNAseq-alignment-genome/featureCounts-gene/featureCounts_gene_matrix.txt.summary \
-o ../output/06.2-cod-RNAseq-alignment-genome/featureCounts-gene
# View directory contents
ls -lh ../output/06.2-cod-RNAseq-alignment-genome/featureCounts-gene
```
I also want to include the treatment/tank info when plotting alignment
rates across samples
``` r
# Load multiqc stats
featureCounts_gene_multiqc <- read.csv("../output/06.2-cod-RNAseq-alignment-genome/featureCounts-gene/multiqc_data/multiqc_featureCounts.txt", sep = '\t')
# Adjust sample name formatting (to prep for join)
featureCounts_gene_multiqc$Sample <- paste("sample_", featureCounts_gene_multiqc$Sample, sep = "")
featureCounts_gene_multiqc_plustreatment <- left_join(cod_sample_info_OG, featureCounts_gene_multiqc, by = c("sample_name" = "Sample")) %>%
na.omit()
featureCounts_gene_multiqc_plustreatment <- featureCounts_gene_multiqc_plustreatment[order(featureCounts_gene_multiqc_plustreatment$sample_number),]
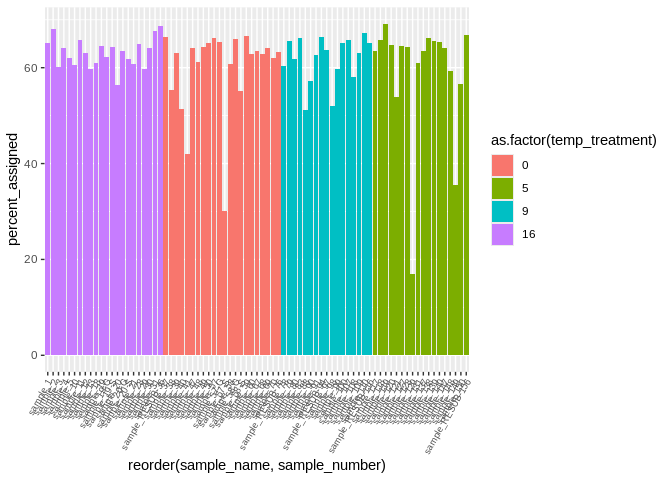
ggplot(featureCounts_gene_multiqc_plustreatment,
aes(x=reorder(sample_name, sample_number), y=percent_assigned, fill=as.factor(temp_treatment))) +
geom_bar(stat="identity") +
theme(axis.text.x = element_text(angle = 60, vjust = 1, hjust=1, size=7))
```
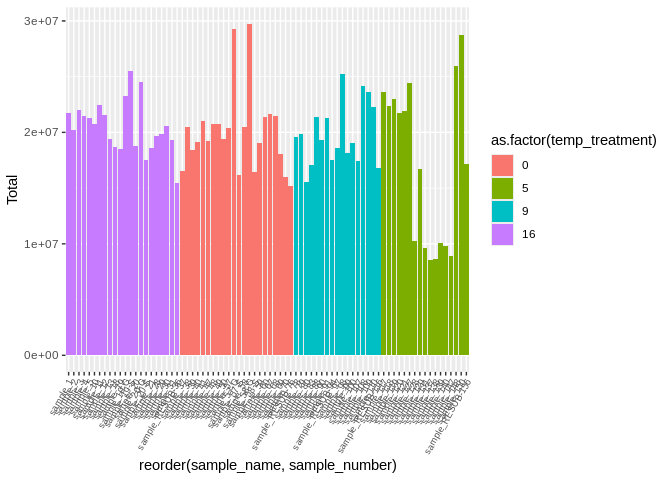
``` r
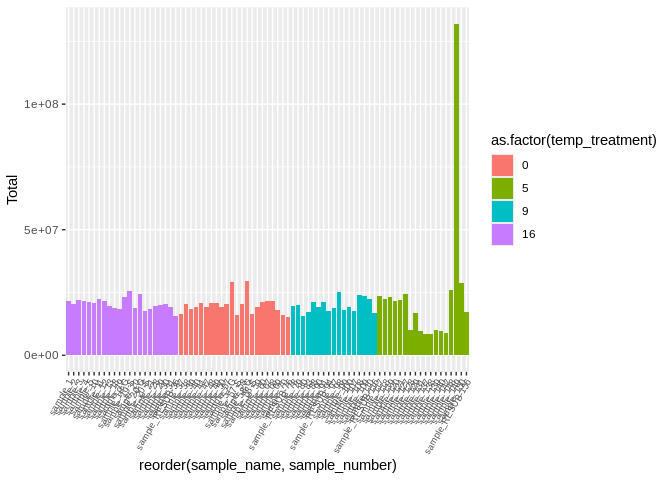
ggplot(featureCounts_gene_multiqc_plustreatment,
aes(x=reorder(sample_name, sample_number), y=Total, fill=as.factor(temp_treatment))) +
geom_bar(stat="identity") +
theme(axis.text.x = element_text(angle = 60, vjust = 1, hjust=1, size=7))
```
``` r
# sample149 is kind of throwing off the visualization, so lets remove and redo
ggplot(featureCounts_gene_multiqc_plustreatment[featureCounts_gene_multiqc_plustreatment$sample_name != "sample_149", ],
aes(x=reorder(sample_name, sample_number), y=Total, fill=as.factor(temp_treatment))) +
geom_bar(stat="identity") +
theme(axis.text.x = element_text(angle = 60, vjust = 1, hjust=1, size=7))
```