R 4.2.3
·
~/GitHub/chris-musselcon/
|
|
|
|
R version 4.2.3 (2023-03-15) -- "Shortstop Beagle" Copyright (C) 2023 The R Foundation for Statistical Computing Platform: x86_64-pc-linux-gnu (64-bit) R is free software and comes with ABSOLUTELY NO WARRANTY. You are welcome to redistribute it under certain conditions. Type 'license()' or 'licence()' for distribution details. Natural language support but running in an English locale R is a collaborative project with many contributors. Type 'contributors()' for more information and 'citation()' on how to cite R or R packages in publications. Type 'demo()' for some demos, 'help()' for on-line help, or 'help.start()' for an HTML browser interface to help. Type 'q()' to quit R. [Workspace loaded from ~/GitHub/chris-musselcon/.RData] > library(knitr) > library(tidyverse) > library(kableExtra) > library(DESeq2) > library(pheatmap) > library(RColorBrewer) > library(data.table) > library(DT) > library(Biostrings) > knitr::opts_chunk$set( + echo = TRUE, # Display code chunks + eval = FALSE, # Evaluate code chunks + warning = FALSE, # Hide warnings + message = FALSE, # Hide messages + fig.width = 6, # Set plot width in inches + fig.height = 4, # Set plot height in inches + fig.align = "center" # Align plots to the center + ) > getwd() [1] "/home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/code" > > > countmatrix <- read.delim("../output/kallisto_01.isoform.counts.matrix", header = TRUE, sep = '\t') > rownames(countmatrix) <- countmatrix$X > countmatrix <- countmatrix[,-1] > head(countmatrix) > #tail(countmatrix) > #dim(countmatrix) > > countmatrix <- round(countmatrix, 0) > str(countmatrix) 'data.frame': 78735 obs. of 8 variables: $ SRR7725722 : num 0 18 0 84 0 56 0 0 0 100 ... $ SRR7725722_1 : num 0 17 0 21 32 49 0 0 0 69 ... $ SRR7725722_2 : num 0 12 0 94 5 49 0 0 0 80 ... $ SRR13013756 : num 2 6 1 33 0 5 0 1 0 0 ... $ SRR16771870 : num 0 2 0 0 0 ... $ SRR16771870_1: num 0 1 1 0 0 ... $ SRR16771870_2: num 0 1 0 0 5 ... $ SRR19782039 : num 0 0 2 0 0 9 0 0 0 295 ... > > > dim(countmatrix) [1] 78735 8 > > dim(deseq2.colData) Error: object 'deseq2.colData' not found > > length(colnames(data)) [1] 0 > > deseq2.colData <- data.frame(condition=factor(c("DSP", "DSP", "DSP", "Hypoxia", "EE2", "EE2", "EE2", "Valsartan")), + type=factor(rep("single-read", 24))) > rownames(deseq2.colData) <- colnames(data) > deseq2.dds <- DESeqDataSetFromMatrix(countData = countmatrix, + colData = deseq2.colData, + design = ~ condition) Error in DESeqDataSetFromMatrix(countData = countmatrix, colData = deseq2.colData, : ncol(countData) == nrow(colData) is not TRUE > countmatrix <- countmatrix[,-1] > head(countmatrix) > countmatrix <- round(countmatrix, 0) > str(countmatrix) 'data.frame': 78735 obs. of 7 variables: $ SRR7725722_1 : num 0 17 0 21 32 49 0 0 0 69 ... $ SRR7725722_2 : num 0 12 0 94 5 49 0 0 0 80 ... $ SRR13013756 : num 2 6 1 33 0 5 0 1 0 0 ... $ SRR16771870 : num 0 2 0 0 0 ... $ SRR16771870_1: num 0 1 1 0 0 ... $ SRR16771870_2: num 0 1 0 0 5 ... $ SRR19782039 : num 0 0 2 0 0 9 0 0 0 295 ... > > > dim(countmatrix) [1] 78735 7 > > #dim(deseq2.colData) > > > length(colnames(data)) [1] 0 > > deseq2.colData <- data.frame(condition=factor(c("DSP", "DSP", "DSP", "Hypoxia", "EE2", "EE2", "EE2", "Valsartan")), + type=factor(rep("single-read", 24))) > rownames(deseq2.colData) <- colnames(data) > deseq2.dds <- DESeqDataSetFromMatrix(countData = countmatrix, + colData = deseq2.colData, + design = ~ condition) Error in DESeqDataSetFromMatrix(countData = countmatrix, colData = deseq2.colData, : ncol(countData) == nrow(colData) is not TRUE Connected to your session in progress, last started 2023-Apr-28 17:41:13 UTC (2 hours ago) > dim(deseq2.colData) [1] 24 2 > deseq2.colData <- data.frame(condition=factor(c("DSP", "Hypoxia", "EE2", "Valsartan")), + type=factor(rep("single-read", 24))) > rownames(deseq2.colData) <- colnames(data) > dim(deseq2.colData) [1] 24 2 > deseq2.dds <- DESeqDataSetFromMatrix(countData = countmatrix, + colData = deseq2.colData, + design = ~ condition) Error in DESeqDataSetFromMatrix(countData = countmatrix, colData = deseq2.colData, : ncol(countData) == nrow(colData) is not TRUE > deseq2.colData <- data.frame(condition=factor(c("DSP", "Hypoxia", "EE2", "Valsartan")), + type=factor(rep("single-read", 8))) > rownames(deseq2.colData) <- colnames(data) > dim(deseq2.colData) [1] 8 2 > deseq2.dds <- DESeqDataSetFromMatrix(countData = countmatrix, + colData = deseq2.colData, + design = ~ condition) Error in DESeqDataSetFromMatrix(countData = countmatrix, colData = deseq2.colData, : ncol(countData) == nrow(colData) is not TRUE > deseq2.colData <- data.frame(condition=factor(c("DSP", "Hypoxia", "EE2", "Valsartan")), + type=factor(rep("single-read", 7))) Error in data.frame(condition = factor(c("DSP", "Hypoxia", "EE2", "Valsartan")), : arguments imply differing number of rows: 4, 7 > deseq2.colData <- data.frame(condition=factor(c("DSP", "DSP", "DSP", "Hypoxia", "EE2", "EE2", "Valsartan")), + type=factor(rep("single-read", 7))) > rownames(deseq2.colData) <- colnames(data) > dim(deseq2.colData) [1] 7 2 > deseq2.dds <- DESeqDataSetFromMatrix(countData = countmatrix, + colData = deseq2.colData, + design = ~ condition) > > deseq2.dds <- DESeq(deseq2.dds) > deseq2.res <- results(deseq2.dds) > deseq2.res <- deseq2.res[order(rownames(deseq2.res)), ] > > > vsd <- vst(deseq2.dds, blind = FALSE) > plotPCA(vsd, intgroup = "condition") > > deseq2.colData <- data.frame(condition=factor(c("DSP", "DSP", "Hypoxia", "EE2", "EE2", "EE2", "Valsartan")), + type=factor(rep("single-read", 7))) > rownames(deseq2.colData) <- colnames(data) > dim(deseq2.colData) [1] 7 2 > deseq2.dds <- DESeqDataSetFromMatrix(countData = countmatrix, + colData = deseq2.colData, + design = ~ condition) > > deseq2.dds <- DESeq(deseq2.dds) > deseq2.res <- results(deseq2.dds) > deseq2.res <- deseq2.res[order(rownames(deseq2.res)), ] > > > vsd <- vst(deseq2.dds, blind = FALSE) > plotPCA(vsd, intgroup = "condition") > > > # Select top 50 differentially expressed genes > res <- results(deseq2.dds) > res_ordered <- res[order(res$padj), ] > top_genes <- row.names(res_ordered)[1:50] > > # Extract counts and normalize > counts <- counts(deseq2.dds, normalized = TRUE) > counts_top <- counts[top_genes, ] > > # Log-transform counts > log_counts_top <- log2(counts_top + 1) > > # Generate heatmap > pheatmap(log_counts_top, scale = "row") > > > head(deseq2.res) log2 fold change (MLE): condition Valsartan vs DSP Wald test p-value: condition Valsartan vs DSP DataFrame with 6 rows and 6 columns baseMean log2FoldChange lfcSE stat pvalue padj <numeric> <numeric> <numeric> <numeric> <numeric> <numeric> lcl|UYJE01000001.1_cds_VDH88688.1_1 30.36796 0.4334722 1.53280 0.2827969 0.777333 0.853160 lcl|UYJE01000001.1_cds_VDH88689.1_2 37.00375 1.6323166 1.60149 1.0192485 0.308085 0.465245 lcl|UYJE01000001.1_cds_VDH88692.1_3 4.90988 -5.0006431 5.39994 -0.9260557 0.354417 NA lcl|UYJE01000001.1_cds_VDH88693.1_4 1.90777 -0.0547951 5.46983 -0.0100177 0.992007 NA lcl|UYJE01000001.1_cds_VDH88694.1_5 22.85891 -5.7625548 3.54438 -1.6258310 0.103986 0.220709 lcl|UYJE01000001.1_cds_VDH88695.1_7 0.00000 NA NA NA NA NA > > > # Count number of hits with adjusted p-value less then 0.05 > dim(deseq2.res[!is.na(deseq2.res$padj) & deseq2.res$padj <= 0.05, ]) [1] 2849 6 > > > tmp <- deseq2.res > # The main plot > plot(tmp$baseMean, tmp$log2FoldChange, pch=20, cex=0.45, ylim=c(-3, 3), log="x", col="darkgray", + main="DEG Valsartan vs DSP (pval <= 0.05)", + xlab="mean of normalized counts", + ylab="Log2 Fold Change") > # Getting the significant points and plotting them again so they're a different color > tmp.sig <- deseq2.res[!is.na(deseq2.res$padj) & deseq2.res$padj <= 0.05, ] > points(tmp.sig$baseMean, tmp.sig$log2FoldChange, pch=20, cex=0.45, col="red") > # 2 FC lines > abline(h=c(-1,1), col="blue") > > > # Prepare the data for plotting > res_df <- as.data.frame(deseq2.res) > res_df$gene <- row.names(res_df) > > # Create volcano plot > volcano_plot <- ggplot(res_df, aes(x = log2FoldChange, y = -log10(padj), color = padj < 0.05)) + + geom_point(alpha = 0.6, size = 1.5) + + scale_color_manual(values = c("grey", "red")) + + labs(title = "Volcano Plot", + x = "Log2 Fold Change", + y = "-Log10 Adjusted P-value", + color = "Significantly\nDifferentially Expressed") + + theme_minimal() + + theme(panel.grid.major = element_blank(), + panel.grid.minor = element_blank(), + legend.position = "top") > > print(volcano_plot) > > > write.table(tmp.sig, "../output/DEGlist.tab", sep = '\t', row.names = T) > > deglist <- read.csv("../output/DEGlist.tab", sep = '\t', header = TRUE) > deglist$RowName <- rownames(deglist) > deglist2 <- deglist[, c("RowName", "pvalue")] # Optionally, reorder the columns > > datatable(deglist) > | ||
|
Console |
|
|
|
|
|
|
|
 Name | Size | Modified |
|---|
.. | ||||
.gitignore | 47 B | Apr 25, 2023, 5:27 PM | ||
.RData | 4.7 MB | Apr 27, 2023, 10:45 PM | ||
.Rhistory | 4.2 KB | Apr 27, 2023, 10:45 PM | ||
applications | ||||
assets | ||||
chris-musselcon.Rproj | 205 B | Apr 28, 2023, 10:41 AM | ||
code | ||||
data | ||||
output | ||||
README.md | 1.1 KB | Apr 13, 2023, 1:36 PM | ||
test file_files | ||||
test file.html | 31.1 KB | Apr 27, 2023, 8:33 PM |
|
|
|
|
81:73 | Chunk 8 | R Markdown |
Downloading the RNASeq data from the accession numbers using SRA Toolkit
Pulling my reference genome from prefab code provided by NCBI
OLD Genome - not needed
Couldn't move unzip here, moved to terminal and did it
Kallisto
Inspect what we have first, then run Kallisto
Create the index file to align my short read files to the genes from the MGAL_10
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
293:1 | Chunk 27 | Quarto |
Pulling my reference genome from prefab code provided by NCBI
Create the index file to align my short read files to the genes from the MGAL_10
Where is data
Runnning Kallisto
DESeq from Kallisto output
trouble calling the program from the whole chunk below so I separated it to make sure I'm going to the right place.
not able to run this without the full path - pretty sure i am missing something there, but this is the solution now.
288
289
290
291
292
293
294
295
296
```
```{r}
datatable(deglist)
```
הההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההההה
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
── Attaching core tidyverse packages ───────────────────────────────────────────────────────────────────── tidyverse 2.0.0 ── ✔ dplyr 1.1.1 ✔ readr 2.1.4 ✔ forcats 1.0.0 ✔ stringr 1.5.0 ✔ ggplot2 3.4.1 ✔ tibble 3.2.1 ✔ lubridate 1.9.2 ✔ tidyr 1.3.0 ✔ purrr 1.0.1 ── Conflicts ─────────────────────────────────────────────────────────────────────────────────────── tidyverse_conflicts() ── ✖ dplyr::filter() masks stats::filter() ✖ dplyr::lag() masks stats::lag() ℹ Use the conflicted package to force all conflicts to become errors Attaching package: ‘kableExtra’ The following object is masked from ‘package:dplyr’: group_rows Loading required package: S4Vectors Loading required package: stats4 Loading required package: BiocGenerics Attaching package: ‘BiocGenerics’ The following objects are masked from ‘package:lubridate’: intersect, setdiff, union The following objects are masked from ‘package:dplyr’: combine, intersect, setdiff, union The following objects are masked from ‘package:stats’: IQR, mad, sd, var, xtabs The following objects are masked from ‘package:base’: anyDuplicated, aperm, append, as.data.frame, basename, cbind, colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find, get, grep, grepl, intersect, is.unsorted, lapply, Map, mapply, match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank, rbind, Reduce, rownames, sapply, setdiff, sort, table, tapply, union, unique, unsplit, which.max, which.min Attaching package: ‘S4Vectors’ The following objects are masked from ‘package:lubridate’: second, second<- The following objects are masked from ‘package:dplyr’: first, rename The following object is masked from ‘package:tidyr’: expand The following objects are masked from ‘package:base’: expand.grid, I, unname Loading required package: IRanges Attaching package: ‘IRanges’ The following object is masked from ‘package:lubridate’: %within% The following objects are masked from ‘package:dplyr’: collapse, desc, slice The following object is masked from ‘package:purrr’: reduce Loading required package: GenomicRanges Loading required package: GenomeInfoDb Loading required package: SummarizedExperiment Loading required package: MatrixGenerics Loading required package: matrixStats Attaching package: ‘matrixStats’ The following object is masked from ‘package:dplyr’: count Attaching package: ‘MatrixGenerics’ The following objects are masked from ‘package:matrixStats’: colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse, colCounts, colCummaxs, colCummins, colCumprods, colCumsums, colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs, colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats, colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds, colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads, colWeightedMeans, colWeightedMedians, colWeightedSds, colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet, rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods, rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps, rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins, rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks, rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars, rowWeightedMads, rowWeightedMeans, rowWeightedMedians, rowWeightedSds, rowWeightedVars Loading required package: Biobase Welcome to Bioconductor Vignettes contain introductory material; view with 'browseVignettes()'. To cite Bioconductor, see 'citation("Biobase")', and for packages 'citation("pkgname")'. Attaching package: ‘Biobase’ The following object is masked from ‘package:MatrixGenerics’: rowMedians The following objects are masked from ‘package:matrixStats’: anyMissing, rowMedians Registered S3 method overwritten by 'data.table': method from print.data.table data.table 1.14.8 using 24 threads (see ?getDTthreads). Latest news: r-datatable.com Attaching package: ‘data.table’ The following object is masked from ‘package:SummarizedExperiment’: shift The following object is masked from ‘package:GenomicRanges’: shift The following object is masked from ‘package:IRanges’: shift The following objects are masked from ‘package:S4Vectors’: first, second The following objects are masked from ‘package:lubridate’: hour, isoweek, mday, minute, month, quarter, second, wday, week, yday, year The following objects are masked from ‘package:dplyr’: between, first, last The following object is masked from ‘package:purrr’: transpose Registered S3 method overwritten by 'htmlwidgets': method from print.htmlwidget tools:rstudio Loading required package: XVector Attaching package: ‘XVector’ The following object is masked from ‘package:purrr’: compact Attaching package: ‘Biostrings’ The following object is masked from ‘package:base’: strsplit
[1] "/home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/code"
>lcl|UYJE01000001.1_cds_VDH88688.1_1 [locus_tag=MGAL_10B017214] [protein=Hypothetical predicted protein] [protein_id=VDH88688.1] [location=complement(join(55975..56224,59152..59365,60239..60337,61332..61522,64535..64608))] [gbkey=CDS]
ATGAATAGAATTACTGATAGGGACTACGACTACTATGACTTTGAAGATGACAGTGACCACGAGCCTTGCGATAGTTCTGA
TGATGATATCGAGGTTATTTTACATGGAACACCTGAACAGAAGCGTAAATTACAGACCAAAGTCCAACAAAGACATGATT
CTTCAAGTGAAGATGACTTTGAAAAGGAAATGAATAATGAACTTAACAAACATATTAAAGGACTGGTAAATGAAAGATCA
AGTAATGTTGCAGAAACTGTTCAAGGTAGTAGCAAAGCTCAAGACCAAGAGAAACCAACAGAACAACAACAATTTTATGA
TGATATTTATTACGATTCAGAAGAAGAGGAAATGGTTTTACAAGGTGATGAACGTGTCAAAAGAAGACAACCTGTTCAAA
GCAATGATGACTTATTGTACGATCCTGACCTAGACGAAGAAGACCAGCGATGGGTTGATGCTGAACGACAAGCTTATCAG
CTGCCTGTACCCTCAGGATCCAAATCAAAACGTCAAAACAGTGATGCAGTTTTAAACTGTCCCGCTTGTATGACATTACT
GTGTCTTGATTGTCAGGGGCATGATGTTTATGAAAACCAGTACAGAGCTATGTTTGTTAAGAACTGTCGTGTCGATACAT
CAGAATTATTAAAACAGCCGTTACAGAAGAAAAAACGTAAAAAAAAACAGAAGACATTGGACACTACAAATAATGAAACA
[build] loading fasta file ../data//ncbi_dataset/data/GCA_900618805.1/cds_from_genomic.fna
[build] k-mer length: 31
[build] warning: replaced 159 non-ACGUT characters in the input sequence
with pseudorandom nucleotides
[build] counting k-mers ... done.
[build] building target de Bruijn graph ... done
[build] creating equivalence classes ... done
[build] target de Bruijn graph has 536288 contigs and contains 60327706 k-mers
SRR13013756.fastq
SRR13013756_fastqc.html
SRR13013756_fastqc.zip
SRR16771870_1.fastq
SRR16771870_1_fastqc.html
SRR16771870_1_fastqc.zip
SRR16771870_2.fastq
SRR16771870_2_fastqc.html
SRR16771870_2_fastqc.zip
SRR19782039.fastq
SRR19782039_fastqc.html
SRR19782039_fastqc.zip
SRR7725722_1.fastq
SRR7725722_1_fastqc.html
SRR7725722_1_fastqc.zip
SRR7725722_2.fastq
SRR7725722_2_fastqc.html
SRR7725722_2_fastqc.zip
total 96079487 avg=92.002677 stddev=405.167227
[quant] fragment length distribution is truncated gaussian with mean = 92, sd = 405
[index] k-mer length: 31
[index] number of targets: 78,735
[index] number of k-mers: 60,327,706
[index] number of equivalence classes: 191,328
[quant] running in single-end mode
[quant] will process file 1: /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/ncbi/SRR13013756.fastq
[quant] finding pseudoalignments for the reads ... done
[quant] processed 8,047,341 reads, 4,102,403 reads pseudoaligned
[ em] quantifying the abundances ... done
[ em] the Expectation-Maximization algorithm ran for 1,028 rounds
[quant] fragment length distribution is truncated gaussian with mean = 92, sd = 405
[index] k-mer length: 31
[index] number of targets: 78,735
[index] number of k-mers: 60,327,706
[index] number of equivalence classes: 191,328
[quant] running in single-end mode
[quant] will process file 1: /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/ncbi/SRR16771870_1.fastq
[quant] finding pseudoalignments for the reads ... done
[quant] processed 19,661,720 reads, 8,025,344 reads pseudoaligned
[ em] quantifying the abundances ... done
[ em] the Expectation-Maximization algorithm ran for 1,304 rounds
[quant] fragment length distribution is truncated gaussian with mean = 92, sd = 405
[index] k-mer length: 31
[index] number of targets: 78,735
[index] number of k-mers: 60,327,706
[index] number of equivalence classes: 191,328
[quant] running in single-end mode
[quant] will process file 1: /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/ncbi/SRR16771870_2.fastq
[quant] finding pseudoalignments for the reads ... done
[quant] processed 19,661,720 reads, 7,631,625 reads pseudoaligned
[ em] quantifying the abundances ... done
[ em] the Expectation-Maximization algorithm ran for 1,095 rounds
[quant] fragment length distribution is truncated gaussian with mean = 92, sd = 405
[index] k-mer length: 31
[index] number of targets: 78,735
[index] number of k-mers: 60,327,706
[index] number of equivalence classes: 191,328
[quant] running in single-end mode
[quant] will process file 1: /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/ncbi/SRR19782039.fastq
[quant] finding pseudoalignments for the reads ... done
[quant] processed 5,564,824 reads, 1,314,760 reads pseudoaligned
[ em] quantifying the abundances ... done
[ em] the Expectation-Maximization algorithm ran for 1,029 rounds
[quant] fragment length distribution is truncated gaussian with mean = 92, sd = 405
[index] k-mer length: 31
[index] number of targets: 78,735
[index] number of k-mers: 60,327,706
[index] number of equivalence classes: 191,328
[quant] running in single-end mode
[quant] will process file 1: /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/ncbi/SRR7725722_1.fastq
[quant] finding pseudoalignments for the reads ... done
[quant] processed 21,571,941 reads, 5,265,435 reads pseudoaligned
[ em] quantifying the abundances ... done
[ em] the Expectation-Maximization algorithm ran for 1,189 rounds
[quant] fragment length distribution is truncated gaussian with mean = 92, sd = 405
[index] k-mer length: 31
[index] number of targets: 78,735
[index] number of k-mers: 60,327,706
[index] number of equivalence classes: 191,328
[quant] running in single-end mode
[quant] will process file 1: /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/ncbi/SRR7725722_2.fastq
[quant] finding pseudoalignments for the reads ... done
[quant] processed 21,571,941 reads, 5,385,311 reads pseudoaligned
[ em] quantifying the abundances ... done
[ em] the Expectation-Maximization algorithm ran for 1,173 rounds
[1] "/home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/code"
-reading file: /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/kallisto_01/SRR7725722/abundance.tsv
-reading file: /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/kallisto_01/SRR7725722_1/abundance.tsv
-reading file: /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/kallisto_01/SRR7725722_2/abundance.tsv
-reading file: /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/kallisto_01/SRR13013756/abundance.tsv
-reading file: /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/kallisto_01/SRR16771870/abundance.tsv
-reading file: /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/kallisto_01/SRR16771870_1/abundance.tsv
-reading file: /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/kallisto_01/SRR16771870_2/abundance.tsv
-reading file: /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/kallisto_01/SRR19782039/abundance.tsv
* Outputting combined matrix.
/home/shared/trinityrnaseq-v2.12.0/util/support_scripts/run_TMM_scale_matrix.pl --matrix /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/kallisto_01.isoform.TPM.not_cross_norm > /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/kallisto_01.isoform.TMM.EXPR.matrixCMD: R --no-save --no-restore --no-site-file --no-init-file -q < /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/kallisto_01.isoform.TPM.not_cross_norm.runTMM.R 1>&2
sh: 1: R: not found
Error, cmd: R --no-save --no-restore --no-site-file --no-init-file -q < /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/kallisto_01.isoform.TPM.not_cross_norm.runTMM.R 1>&2 died with ret (32512) at /home/shared/trinityrnaseq-v2.12.0/util/support_scripts/run_TMM_scale_matrix.pl line 105.
Error, CMD: /home/shared/trinityrnaseq-v2.12.0/util/support_scripts/run_TMM_scale_matrix.pl --matrix /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/kallisto_01.isoform.TPM.not_cross_norm > /home/shared/8TB_HDD_02/cnmntgna/GitHub/chris-musselcon/output/kallisto_01.isoform.TMM.EXPR.matrix died with ret 6400 at /home/shared/trinityrnaseq-v2.12.0/util/abundance_estimates_to_matrix.pl line 385.
Description:df [6 × 7]
| ABCDEFGHIJ0123456789 |
SRR7725722_1 <dbl> | SRR7725722_2 <dbl> | SRR13013756 <dbl> | SRR16771870 <dbl> | ||
|---|---|---|---|---|---|
| lcl|UYJE01002447.1_cds_VDI10805.1_18417 | 0 | 0 | 2 | 0 | |
| lcl|UYJE01005815.1_cds_VDI40612.1_42956 | 17 | 12 | 6 | 2 | |
| lcl|UYJE01010339.1_cds_VDI82332.1_76960 | 0 | 0 | 1 | 0 | |
| lcl|UYJE01003049.1_cds_VDI16160.1_22882 | 21 | 94 | 33 | 0 | |
| lcl|UYJE01005517.1_cds_VDI37871.1_40750 | 32 | 5 | 0 | 0 | |
| lcl|UYJE01006022.1_cds_VDI42419.1_44451 | 49 | 49 | 5 | 8 |
6 rows | 1-5 of 7 columns
'data.frame': 78735 obs. of 7 variables:
$ SRR7725722_1 : num 0 17 0 21 32 49 0 0 0 69 ...
$ SRR7725722_2 : num 0 12 0 94 5 49 0 0 0 80 ...
$ SRR13013756 : num 2 6 1 33 0 5 0 1 0 0 ...
$ SRR16771870 : num 0 2 0 0 0 ...
$ SRR16771870_1: num 0 1 1 0 0 ...
$ SRR16771870_2: num 0 1 0 0 5 ...
$ SRR19782039 : num 0 0 2 0 0 9 0 0 0 295 ...
[1] 78735 7
[1] 0
[1] 7 2
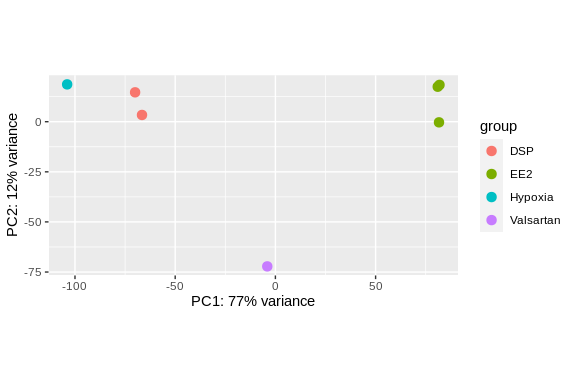
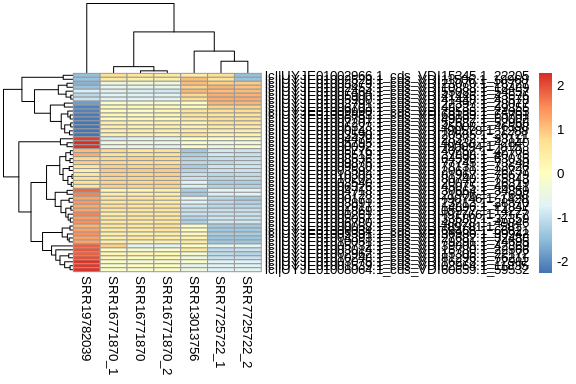
log2 fold change (MLE): condition Valsartan vs DSP
Wald test p-value: condition Valsartan vs DSP
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue padj
<numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
lcl|UYJE01000001.1_cds_VDH88688.1_1 30.36796 0.4334722 1.53280 0.2827969 0.777333 0.853160
lcl|UYJE01000001.1_cds_VDH88689.1_2 37.00375 1.6323166 1.60149 1.0192485 0.308085 0.465245
lcl|UYJE01000001.1_cds_VDH88692.1_3 4.90988 -5.0006431 5.39994 -0.9260557 0.354417 NA
lcl|UYJE01000001.1_cds_VDH88693.1_4 1.90777 -0.0547951 5.46983 -0.0100177 0.992007 NA
lcl|UYJE01000001.1_cds_VDH88694.1_5 22.85891 -5.7625548 3.54438 -1.6258310 0.103986 0.220709
lcl|UYJE01000001.1_cds_VDH88695.1_7 0.00000 NA NA NA NA NA
[1] 2849 6
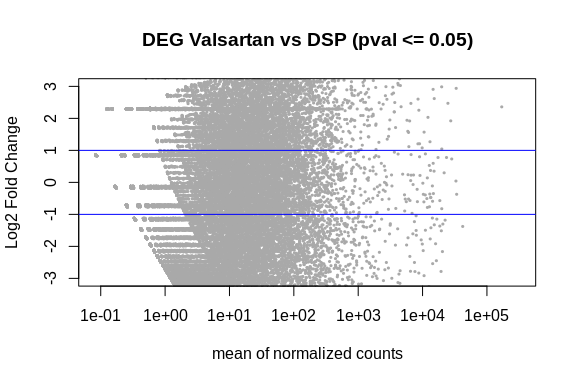
.png)
262:8 | (Top Level) | R Markdown |
```
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
Source |
|
|
|
|
|
Traceback
| Data | |||
| countmatrix | 78735 obs. of 7 variables | ||
| counts | Large matrix (551145 elements, 12.6 MB) | ||
| counts_top | num [1:50, 1:7] 0 7.88 1.82 7.28 44110.87 ... | ||
| deglist | 2849 obs. of 7 variables | ||
| deglist2 | 2849 obs. of 2 variables | ||
| deseq2.colData | 7 obs. of 2 variables | ||
| deseq2.dds | Large DESeqDataSet (78735 elements, 50.1 MB) | ||
| deseq2.res | Large DESeqResults (6 elements, 12 MB) | ||
| log_counts_top | num [1:50, 1:7] 0 3.15 1.5 3.05 15.43 ... | ||
| res | Large DESeqResults (6 elements, 12 MB) | ||
| res_df | 78735 obs. of 7 variables | ||
| res_ordered | Large DESeqResults (6 elements, 12 MB) | ||
| tmp | Large DESeqResults (6 elements, 12 MB) | ||
| tmp.sig | Formal class DESeqResults | ||
| volcano_plot | Large gg (9 elements, 20.2 MB) | ||
| vsd | Large DESeqTransform (78735 elements, 39.1 MB) | ||
| Values | |||
| top_genes | chr [1:50] "lcl|UYJE01003075.1_cds_VDI16278.1_22982" "lcl|UYJE01004718.1_cds_VDI30664.1_34859" "lcl|UYJE01010303.1_cds_VDI81967.1_76679" "lcl|UYJE01010202.1_cds_VDI80740.1_75843" "lcl|UYJE0100590 <...> | ||
| Staged | Status | Path |
|---|---|---|
test file.html | ||
test file_files/ | ||
code/05-presentation_files/ | ||
code/GCA_900618805.1.zip | ||
code/rsconnect/ | ||
data/GCA_025277285.1.zip | ||
data/GCA_900618805.1.zip | ||
data/ncbi_dataset/ | ||
Your branch is ahead of 'origin/main' by 2 commits. | Your branch is ahead of 'origin/main' by 2 commits. |
|