Input data and parameters
QualiMap command line
| qualimap bamqc -bam /Users/strigg/Desktop/20200427/Sealice_F2_S22.dedup.sorted.bam -nw 400 -hm 3 |
Alignment
| Command line: | "bismark --path_to_bowtie /gscratch/srlab/programs/bowtie2-2.3.4.1-linux-x86_64 --samtools_path /gscratch/srlab/programs/samtools-1.9 --score_min L,0,-0.6 -p 4 --non_directional --dovetail --genome /gscratch/srlab/strigg/data/Caligus/GENOMES -1 /gscratch/scrubbed/strigg/TRIMG_adapt_5bp/TRIM_cat/Sealice_F2_S22_R1_001_val_1.fq.gz -2 /gscratch/scrubbed/strigg/TRIMG_adapt_5bp/TRIM_cat/Sealice_F2_S22_R2_001_val_2.fq.gz -o /gscratch/scrubbed/strigg/analyses/20190814_Calig" |
| Draw chromosome limits: | no |
| Analyze overlapping paired-end reads: | no |
| Program: | Bismark (v0.19.0) |
| Analysis date: | Mon Apr 27 13:22:02 PDT 2020 |
| Size of a homopolymer: | 3 |
| Skip duplicate alignments: | no |
| Number of windows: | 400 |
| BAM file: | /Users/strigg/Desktop/20200427/Sealice_F2_S22.dedup.sorted.bam |
Summary
Globals
| Reference size | 506,970,227 |
| Number of reads | 53,248,360 |
| Mapped reads | 53,248,360 / 100% |
| Unmapped reads | 0 / 0% |
| Mapped paired reads | 53,248,360 / 100% |
| Mapped reads, first in pair | 26,624,180 / 50% |
| Mapped reads, second in pair | 26,624,180 / 50% |
| Mapped reads, both in pair | 53,248,360 / 100% |
| Mapped reads, singletons | 0 / 0% |
| Read min/max/mean length | 20 / 145 / 117.35 |
| Duplicated reads (estimated) | 18,562,236 / 34.86% |
| Duplication rate | 24.88% |
| Clipped reads | 0 / 0% |
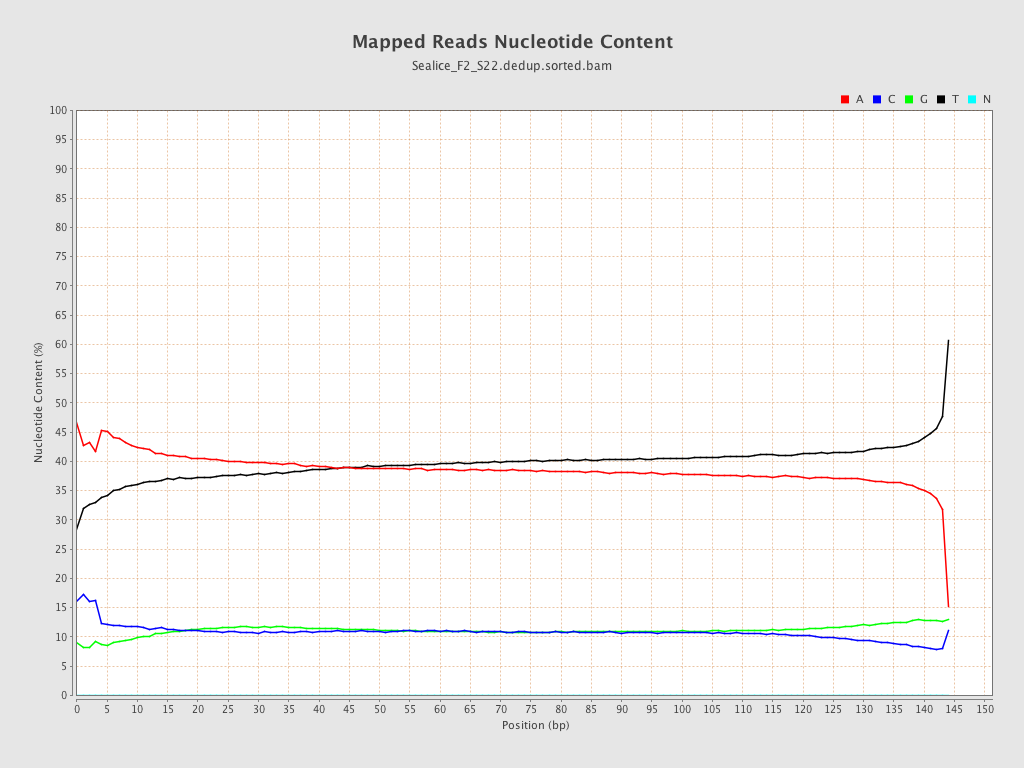
ACGT Content
| Number/percentage of A's | 2,410,240,743 / 39.04% |
| Number/percentage of C's | 675,087,412 / 10.94% |
| Number/percentage of T's | 2,409,933,267 / 39.04% |
| Number/percentage of G's | 677,962,431 / 10.98% |
| Number/percentage of N's | 43,660 / 0% |
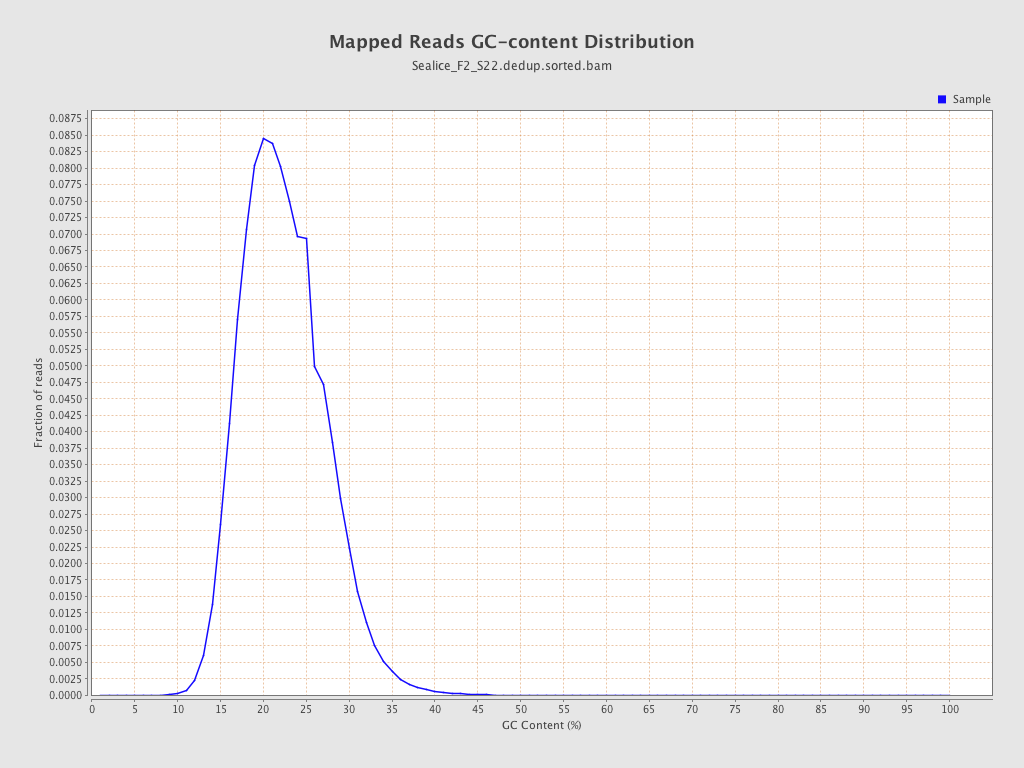
| GC Percentage | 21.92% |
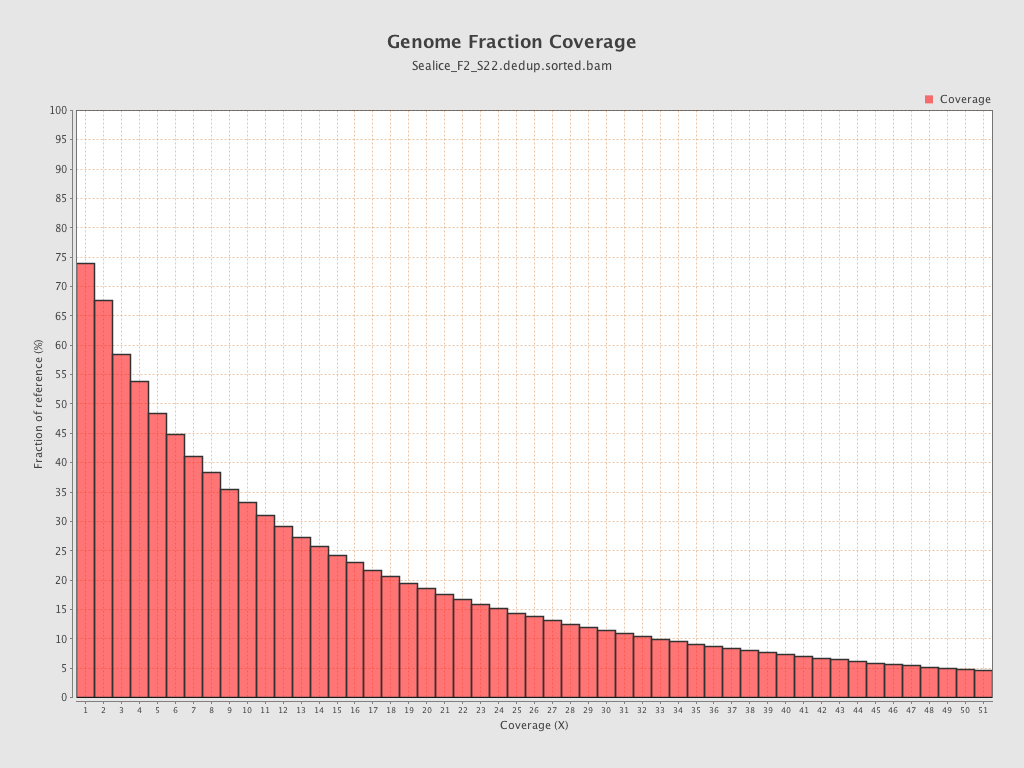
Coverage
| Mean | 12.2283 |
| Standard Deviation | 42.6626 |
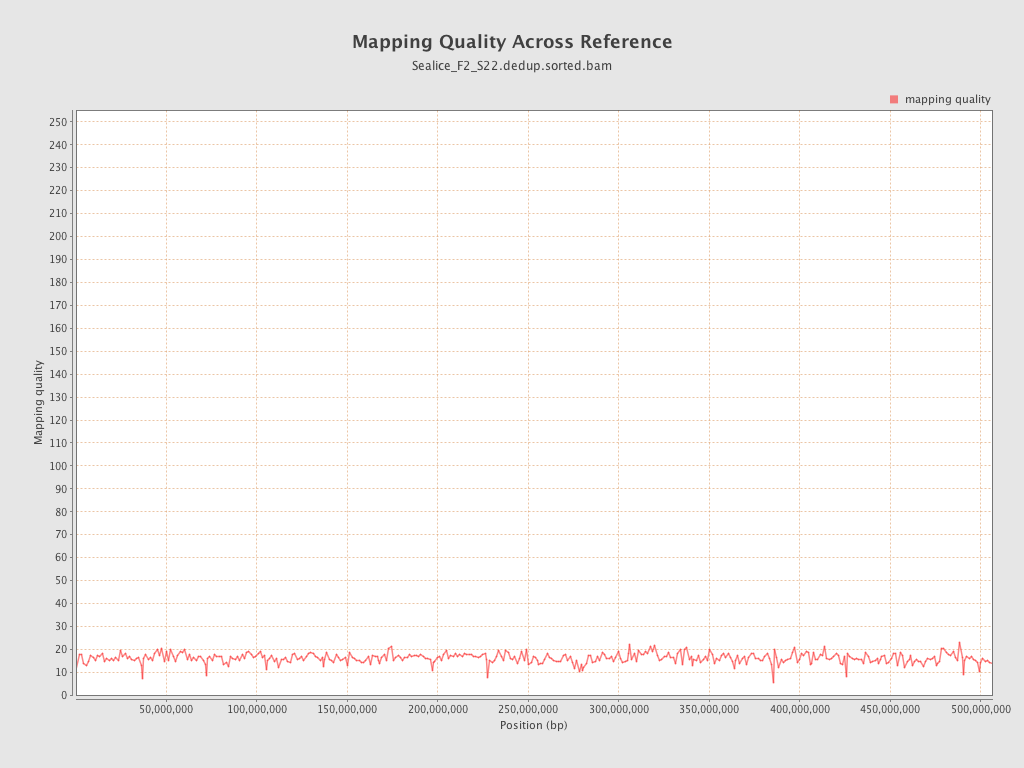
Mapping Quality
| Mean Mapping Quality | 16.13 |
Insert size
| Mean | 142.21 |
| Standard Deviation | 63.74 |
| P25/Median/P75 | 98 / 133 / 169 |
Mismatches and indels
| General error rate | 22.9% |
| Mismatches | 1,343,814,679 |
| Insertions | 38,168,816 |
| Mapped reads with at least one insertion | 45.32% |
| Deletions | 15,516,752 |
| Mapped reads with at least one deletion | 23.86% |
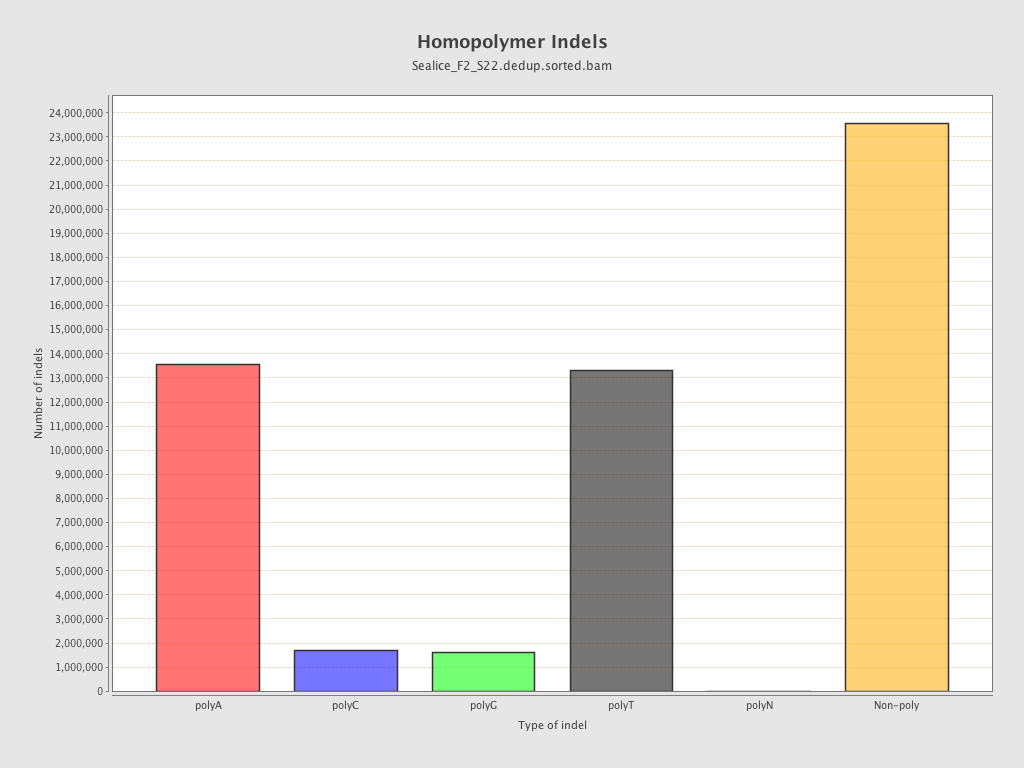
| Homopolymer indels | 56.14% |
Chromosome stats
| Name | Length | Mapped bases | Mean coverage | Standard deviation |
| Cr_scaffold0000001 | 36897007 | 464506567 | 12.5893 | 37.9408 |
| Cr_scaffold0000002 | 35124817 | 413185688 | 11.7634 | 34.4861 |
| Cr_scaffold0000003 | 33138466 | 405102476 | 12.2245 | 34.5681 |
| Cr_scaffold0000004 | 31456035 | 405661622 | 12.8961 | 34.1439 |
| Cr_scaffold0000005 | 30612693 | 363314193 | 11.8681 | 27.7143 |
| Cr_scaffold0000006 | 30259679 | 368168819 | 12.167 | 27.3907 |
| Cr_scaffold0000007 | 29872177 | 349932184 | 11.7143 | 27.6391 |
| Cr_scaffold0000008 | 27803016 | 337074794 | 12.1237 | 72.7837 |
| Cr_scaffold0000009 | 25126635 | 301755889 | 12.0094 | 31.2016 |
| Cr_scaffold0000010 | 24815513 | 299448975 | 12.067 | 28.0814 |
| Cr_scaffold0000011 | 35794548 | 474026835 | 13.243 | 24.8935 |
| Cr_scaffold0000012 | 22957860 | 253685098 | 11.05 | 20.898 |
| Cr_scaffold0000013 | 21502856 | 244210870 | 11.3571 | 22.2185 |
| Cr_scaffold0000014 | 21148584 | 256212990 | 12.1149 | 40.1064 |
| Cr_scaffold0000015 | 19091682 | 239034545 | 12.5204 | 59.3004 |
| Cr_scaffold0000016 | 18525937 | 222909213 | 12.0323 | 66.2656 |
| Cr_scaffold0000017 | 17562062 | 225440852 | 12.8368 | 96.9178 |
| Cr_scaffold0000018 | 14680182 | 171115564 | 11.6562 | 37.4579 |
| Cr_scaffold0000019 | 14290452 | 199010886 | 13.9261 | 66.9634 |
| Cr_scaffold0000020 | 8286978 | 97530248 | 11.7691 | 32.9691 |
| Cr_scaffold0000021 | 8023048 | 108066280 | 13.4695 | 29.6646 |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}