Input data and parameters
QualiMap command line
| qualimap bamqc -bam /Users/strigg/Desktop/20200427/Sealice_F1_S20.dedup.sorted.bam -nw 400 -hm 3 |
Alignment
| Command line: | "bismark --path_to_bowtie /gscratch/srlab/programs/bowtie2-2.3.4.1-linux-x86_64 --samtools_path /gscratch/srlab/programs/samtools-1.9 --score_min L,0,-0.6 -p 4 --non_directional --dovetail --genome /gscratch/srlab/strigg/data/Caligus/GENOMES -1 /gscratch/scrubbed/strigg/TRIMG_adapt_5bp/TRIM_cat/Sealice_F1_S20_R1_001_val_1.fq.gz -2 /gscratch/scrubbed/strigg/TRIMG_adapt_5bp/TRIM_cat/Sealice_F1_S20_R2_001_val_2.fq.gz -o /gscratch/scrubbed/strigg/analyses/20190814_Calig" |
| Draw chromosome limits: | no |
| Analyze overlapping paired-end reads: | no |
| Program: | Bismark (v0.19.0) |
| Analysis date: | Mon Apr 27 13:16:26 PDT 2020 |
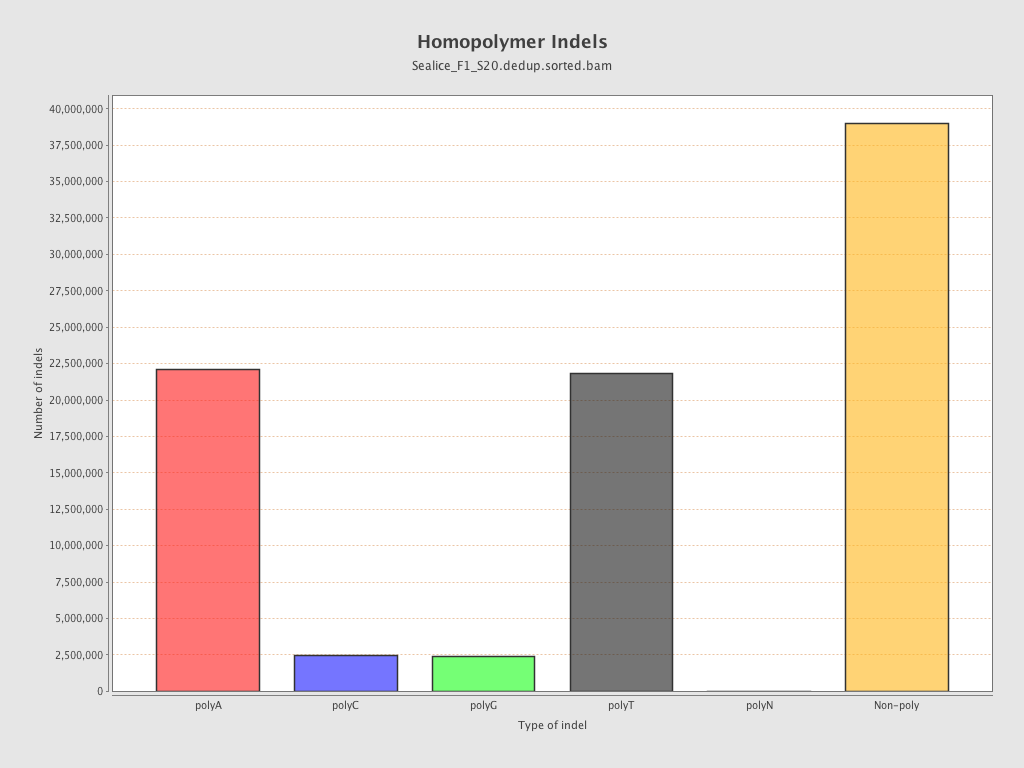
| Size of a homopolymer: | 3 |
| Skip duplicate alignments: | no |
| Number of windows: | 400 |
| BAM file: | /Users/strigg/Desktop/20200427/Sealice_F1_S20.dedup.sorted.bam |
Summary
Globals
| Reference size | 506,970,227 |
| Number of reads | 79,611,238 |
| Mapped reads | 79,611,238 / 100% |
| Unmapped reads | 0 / 0% |
| Mapped paired reads | 79,611,238 / 100% |
| Mapped reads, first in pair | 39,805,619 / 50% |
| Mapped reads, second in pair | 39,805,619 / 50% |
| Mapped reads, both in pair | 79,611,238 / 100% |
| Mapped reads, singletons | 0 / 0% |
| Read min/max/mean length | 20 / 145 / 120.5 |
| Duplicated reads (estimated) | 28,313,082 / 35.56% |
| Duplication rate | 26.13% |
| Clipped reads | 0 / 0% |
ACGT Content
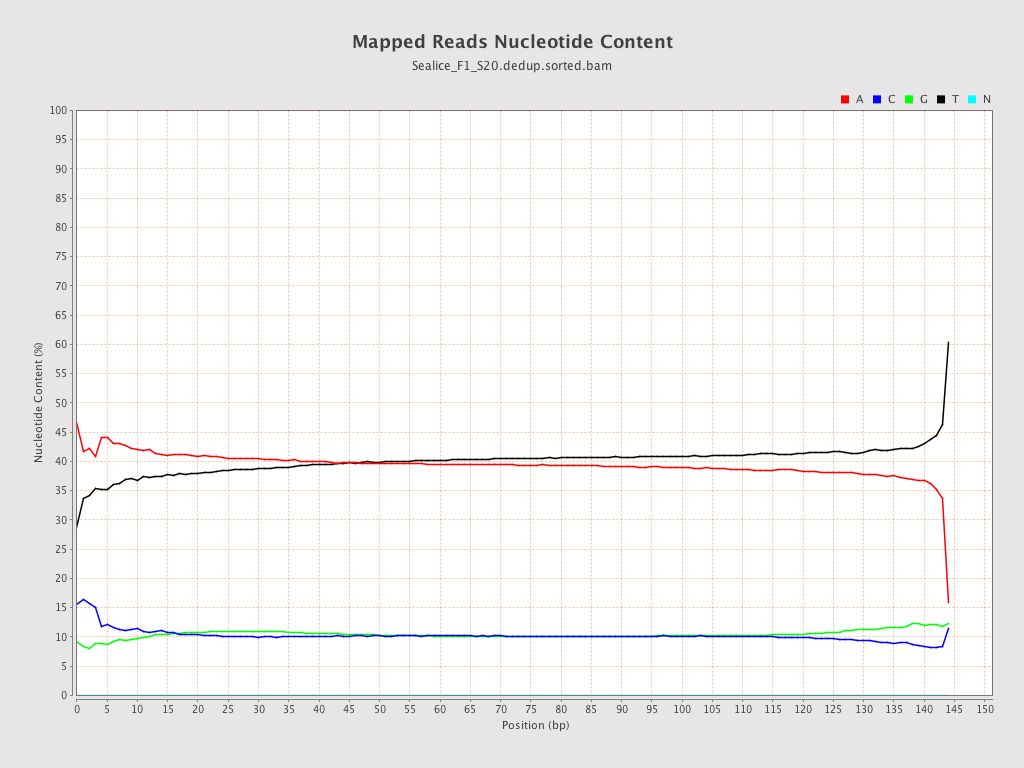
| Number/percentage of A's | 3,762,018,572 / 39.7% |
| Number/percentage of C's | 975,929,878 / 10.3% |
| Number/percentage of T's | 3,759,461,175 / 39.68% |
| Number/percentage of G's | 977,805,541 / 10.32% |
| Number/percentage of N's | 65,145 / 0% |
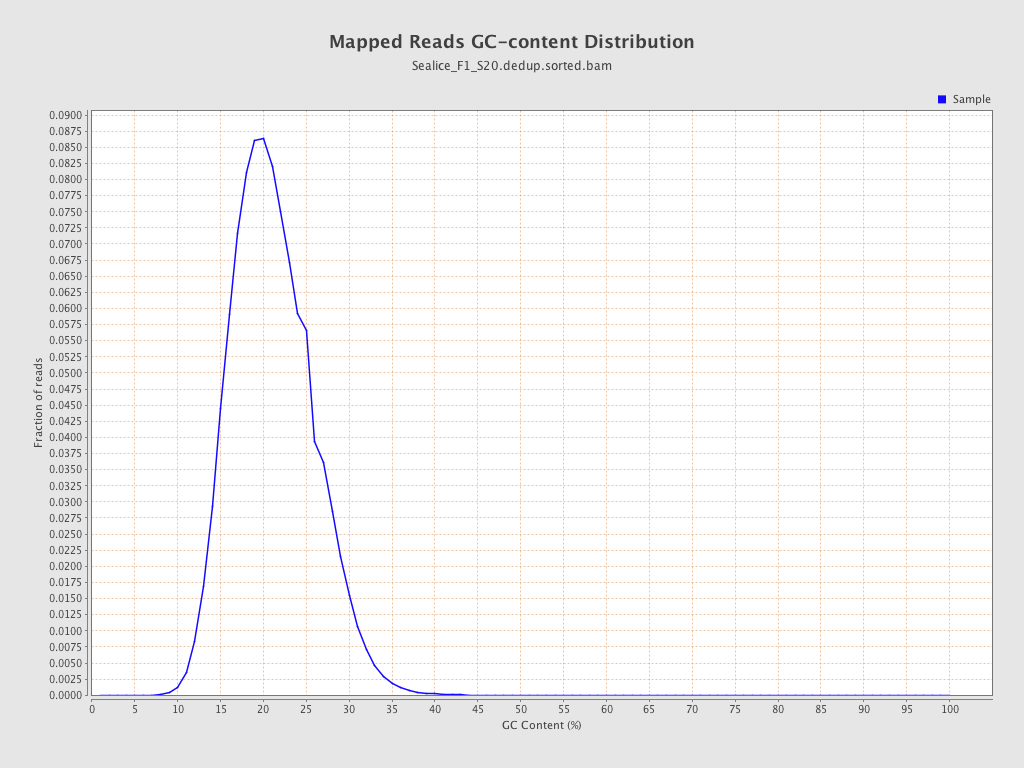
| GC Percentage | 20.62% |
Coverage
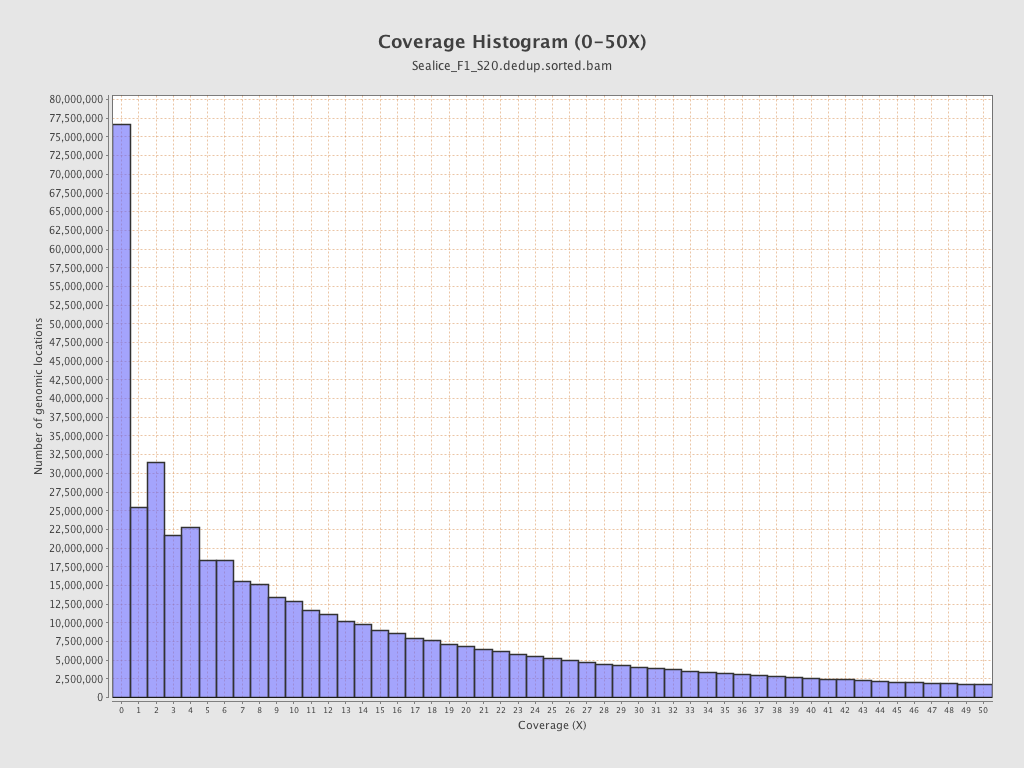
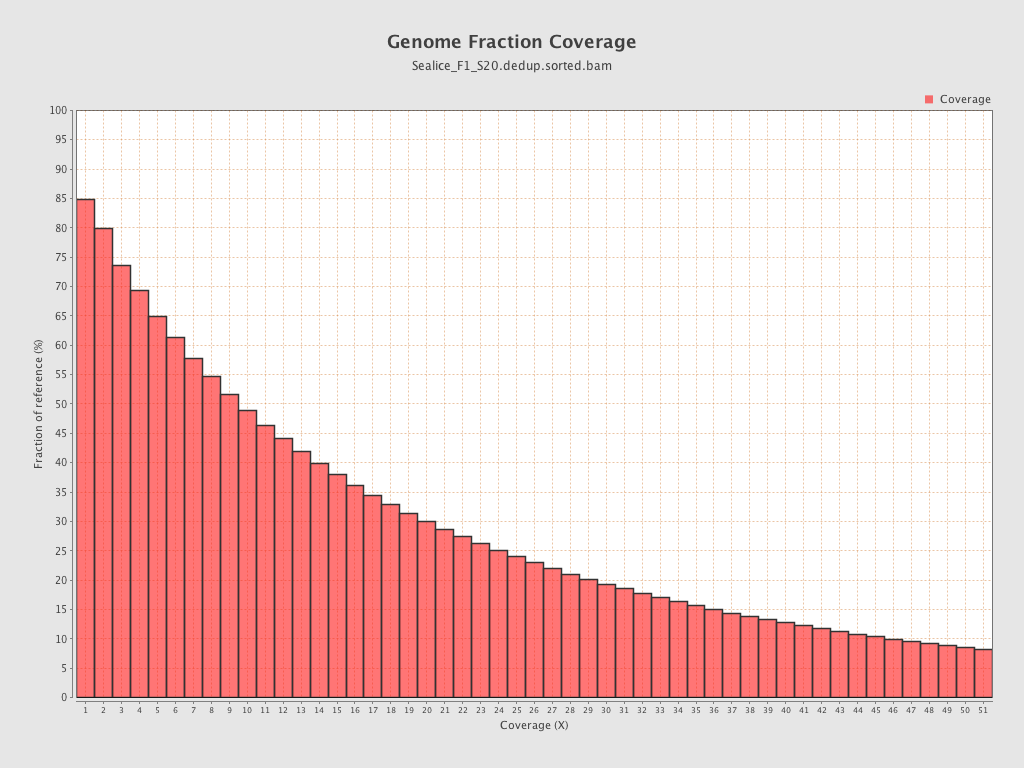
| Mean | 18.7775 |
| Standard Deviation | 66.3745 |
Mapping Quality
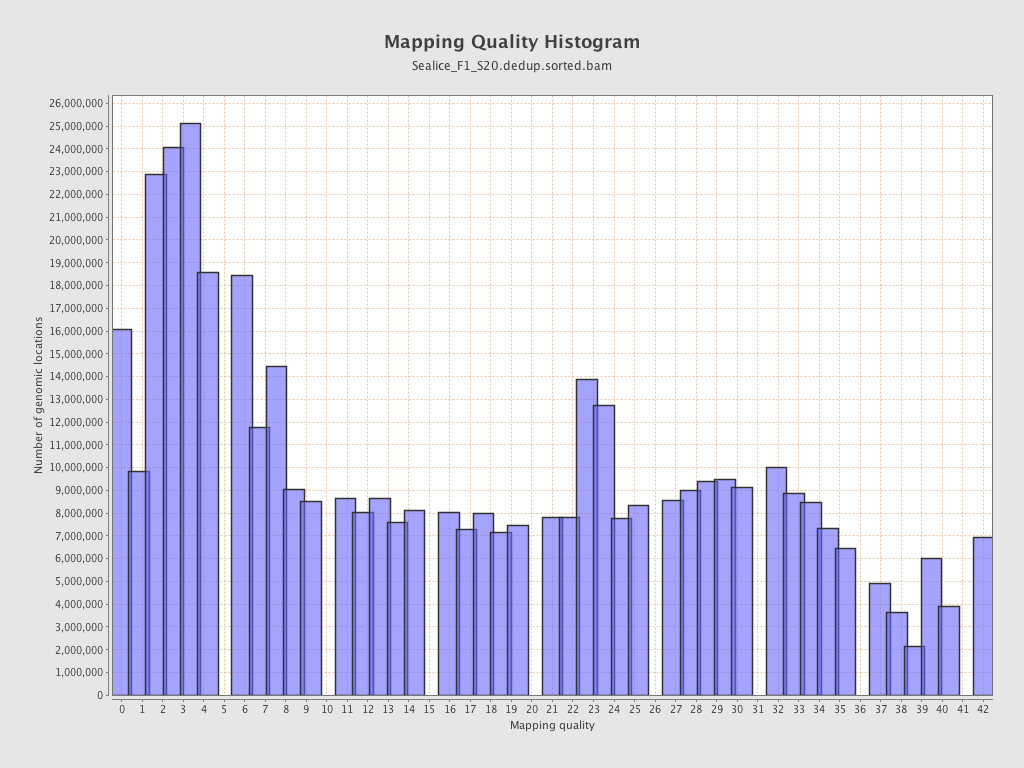
| Mean Mapping Quality | 15.85 |
Insert size
| Mean | 156.9 |
| Standard Deviation | 77.14 |
| P25/Median/P75 | 103 / 143 / 191 |
Mismatches and indels
| General error rate | 22.37% |
| Mismatches | 2,010,625,717 |
| Insertions | 61,265,857 |
| Mapped reads with at least one insertion | 47.51% |
| Deletions | 26,451,178 |
| Mapped reads with at least one deletion | 26.85% |
| Homopolymer indels | 55.57% |
Chromosome stats
| Name | Length | Mapped bases | Mean coverage | Standard deviation |
| Cr_scaffold0000001 | 36897007 | 714865995 | 19.3746 | 61.5053 |
| Cr_scaffold0000002 | 35124817 | 641967240 | 18.2767 | 58.7536 |
| Cr_scaffold0000003 | 33138466 | 627780647 | 18.9442 | 48.563 |
| Cr_scaffold0000004 | 31456035 | 618598046 | 19.6655 | 45.7081 |
| Cr_scaffold0000005 | 30612693 | 559909539 | 18.2901 | 37.2556 |
| Cr_scaffold0000006 | 30259679 | 562698673 | 18.5957 | 37.7551 |
| Cr_scaffold0000007 | 29872177 | 543795664 | 18.2041 | 49.7049 |
| Cr_scaffold0000008 | 27803016 | 528560298 | 19.0109 | 119.7617 |
| Cr_scaffold0000009 | 25126635 | 464855308 | 18.5005 | 44.8314 |
| Cr_scaffold0000010 | 24815513 | 459153774 | 18.5027 | 40.7954 |
| Cr_scaffold0000011 | 35794548 | 716142100 | 20.007 | 33.1539 |
| Cr_scaffold0000012 | 22957860 | 391884456 | 17.0697 | 27.2087 |
| Cr_scaffold0000013 | 21502856 | 375800495 | 17.4768 | 28.6549 |
| Cr_scaffold0000014 | 21148584 | 396498264 | 18.7482 | 59.9442 |
| Cr_scaffold0000015 | 19091682 | 364697101 | 19.1024 | 89.1483 |
| Cr_scaffold0000016 | 18525937 | 336209263 | 18.148 | 50.1623 |
| Cr_scaffold0000017 | 17562062 | 348705035 | 19.8556 | 185.5172 |
| Cr_scaffold0000018 | 14680182 | 266125712 | 18.1282 | 61.6275 |
| Cr_scaffold0000019 | 14290452 | 295773645 | 20.6973 | 99.7446 |
| Cr_scaffold0000020 | 8286978 | 146920877 | 17.7291 | 41.5526 |
| Cr_scaffold0000021 | 8023048 | 158696419 | 19.7801 | 40.9665 |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}